Deviations
In addition to the definition of standard production procedures, it needs to be defined in advance how deviations should be dealt with. This approach ensures that where unforeseen events occur, an acceptable product quality is maintained.
Here you will find the answers to the following questions:
|
11.K.1 Definition
It is important to describe how the term deviation should be interpreted in order to avoid a lack of clarity and subsequent misunderstandings about the procedure and responsibilities involved. As a rule, a deviation has occurred if specifications are not complied with during in-process tests or release analyses (OOS results) (see chapter 14.H Out-of-specification results). Deviations also include process parameters or operating procedures outside the specifications of manufacturing instructions as well as all notable aspects and observations of processes, their statuses and equipment. In this case, the deviation may be regarded as an umbrella term under which OOS results can be included. This grading and specification of the associated responsibilities must be tailored to the specific company. Systems also exist where the term deviations applies exclusively to the production process and in-process data. While this is possible, it does require the handling of OOS results and other deviations to be coordinated jointly.
| Definition of deviations |
|---|
Non-compliance with:
|
11.K.2 Sequence
Once a deviation has occurred, a standard procedure that is defined in terms of a standard process instruction must be initiated for all areas. It is important to determine who the decision makers will be and the degree of flexibility they have when making decisions, particularly as speedy decisions occasionally need to be made about the processing of intermediate products.
Until it is clear how to proceed in the event of a quality deviation, it must be ensured that further processing of the batch concerned is not possible (see chapter 14.H Out-of-specification results). This may be achieved for example by using the EDP system that monitors the individual production steps to quarantine the material (if such a system is used). The material must be labelled in every case (see chapter 11.H Identification) and stored separately. "Rejected materials and products should be clearly marked as such and stored separately in restricted areas." (5.61 EU GMP Guideline).
If the deviation affects controllable parameters, e. g. temperatures or filling quantities, attempts at modification are made to achieve a result that satisfies the requirement through application of the permissible method. Such trials are to be documented. To this end it must be established where this should be carried out. In every case, it must be possible to find proof of these in the batch record. Other forms of documentation are discussed below which make it possible to critically observe the possible consequences during subsequent processing steps. Non-controllable parameters, e. g. discolouration at the end of a process must be fully documented and made available to the decision maker concerned. Processing must then be suspended by the head of production until approval is obtained.
In general, standard elements that are arranged chronologically as part of every procedure for the handling of deviations are to be named. These must be documented individually.
Description of the deviation
The type and scope of the deviation must be described as precisely as possible. In doing so, information on the context must be recorded in order to make retrospective evaluation possible.
Researching causes
Whenever possible, the likely causes of the deviation should be recorded together with its description. Sufficient information must be obtained in order to carry out a conclusive evaluation.
Definition of measures
In order to determine how to proceed, measures must be defined based on a risk analysis - direct measures for the affected batch (in the case of a batch-related deviation) in order to achieve the necessary quality (corrective action) and also more far-reaching measures that prevent reoccurrence in subsequent batches or processes (preventive action). This concept is referred to as CAPA.
| Elements of procedures for the handling of deviations |
|---|
|
In order to obtain sufficient transparency of information and the information chain, these are normally compiled in a failure investigation report. Figure 11.K-3 shows an example of process structuring for the handling of deviations. Information about a deviation is provided by the relevant department
|
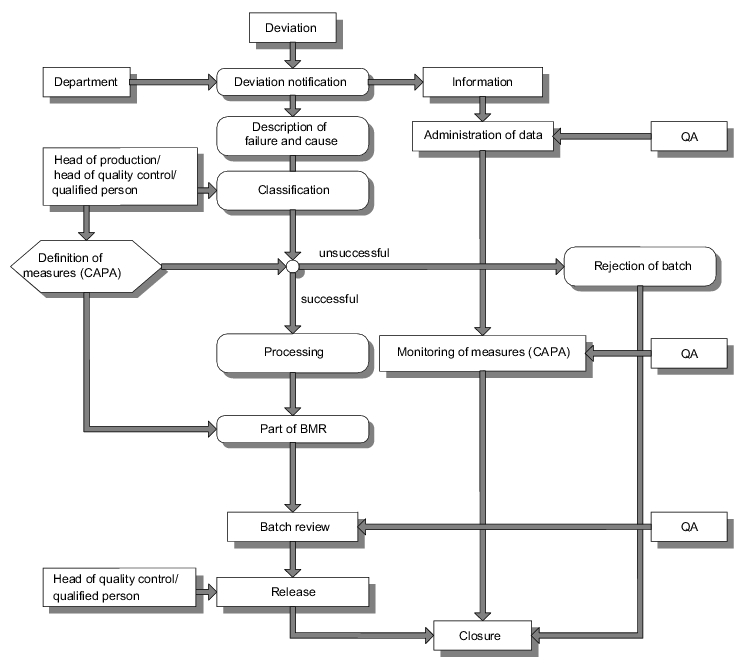
.
Classification of the deviations is recommended (e. g. minor, major, critical), in order to achieve a certain degree of standardisation from the outset as regards the type of measures to be taken. If the measures are not successful, this may lead to the quarantining of the batch, depending on the risk evaluation. Once manufacturing is complete and all information including the failure investigation report is available, the batch is released by the Head of Quality Control/Qualified Person. The status of corrective and preventive action (CAPA) is monitored in this example by quality assurance.
11.K.3 Responsibilities
The decision maker must be clearly identified for cases where a product or intermediate product does not conform with the specifications. For practical handling purposes, it is advantageous to allocate responsibilities in such a way that long standing times can be avoided. This means that it is expedient to appoint persons in specific positions who can act as the decision makers in the first instance and can be reached quickly and at any time, e. g. area manager. Relevant qualifications are prerequisites. The information should be forwarded to the head of manufacturing at an early stage as well as the functions involved in the investigation of causes. These could for example be centres or departments whose task is to evaluate Product Reviews (see chapter 15.F Annual product review). The information must also be made available to the head of quality control or the Qualified Person as this must be taken into account when evaluating the quality of the product. It is therefore recommended that the failure investigation report should be an integral part of the batch documentation (BMR). By the same token, paper-free information and data systems may be used. These systems must be validated in every case as this is an area that is critical from the point of view of Good Manufacturing Practice.
11.K.4 Measures
As described above, a differentiation is made between measures that are applied directly for the relevant batch or specific deviation (immediate measures) and longer term measures that are designed to prevent recurrence or improve processes.
Immediate corrective measures must be authorised by the head of manufacturing during manufacture prior to processing, e. g.:
- 100-percent control for rejection of defective units
- Reworking of the defective product
- Destruction of the defective product
- For starting materials: restricted product-specific approvals, if this can be guaranteed by the approval system; use following the modification of process steps (e. g. coarse preliminary homogenisation of agglomerated substances)
The decision as to the type of measures to be taken is based on a risk analysis and, as far as possible, available data and experiences with the processes. In doing so, compliance with registration-related specifications and validated areas must be ensured.
Depending on the type of deviation, it may be necessary to apply measures across all batches, e. g.:
- Intensified sampling during subsequent manufacturing
- Trend analyses, product review
- Staff training
- Checks on equipment and machines
If process checks reveal that changes to processes or specifications are necessary, in-house change control procedures (see chapter 19.C Change control) must be complied with. For example, specifications for active pharmaceutical ingredients and excipients may need to be further restricted if manufacturing problems are caused by characteristics that, while conforming with the specifications, are at the tolerance limit.
11.K.5 Failure investigation report
All information pertaining to the deviation must be documented in the failure investigation report, i.e. description of the deviation, causes and the measures derived. It is expedient to define a period (e. g. 30 days) within which the report must be compiled. In every case, the information must be available to the head of quality control or the Qualified Person so that it can be taken into account for the batch approval decision. It is recommended that this document is submitted for approval with the batch record as this contains all data relevant to the approval, e. g. from the production area.
It is expedient to number the failure investigation reports and therefore also the deviations - this guarantees that they will be clearly identified. This may be achieved (for example) by using forms that have been numbered beforehand or by creating a special numbering system. References to these numbers should be provided at defined positions in the investigation reports or batch production records.
|

Out-of-specification quality control results (OOS) lead to an investigation of the manufacturing process once a laboratory error has been excluded (see chapter 14.H Out-of-specification results). The appraisal of the batch processing record must initially be carried out, i.e. what is the extent of the deviation in relation to the specified parameters. If no deviations are found, the relevant batch as well as the previous batches and validation batches are analysed retrospectively. Evaluations of log books, staff surveys and comparisons of starting material batches used to date are used for the assessment. This information is consolidated with secondary documentation such as environmental control values. Similar to the investigation in the laboratory area, it is recommended that checklists are also used here when performing the reviews.
11.K.6 Evaluation
The implementation and effectiveness of the measures taken following the deviation and failure investigation must be checked. To this end, various responsibilities can be assigned according to specific company requirements. In the example shown here, this is carried out by quality assurance which is informed about the deviation as soon as it is found. The effectiveness of the measures taken can then be checked during the manufacturing process when reviewing the batch document for example.
| Data fields for the administration of deviations |
|---|
|
It is recommended that all deviations are managed using a database (see figure 11.K-5). In doing so, a failure classification system should be compiled that makes it possible to perform a detailed breakdown according to failure categories. Statistical evaluation of the information registered makes possible critical process- and product-specific assessment of process elements which in turn uncovers trends. By recording the status of specified measures, timely compliance with the specifications can be regularly checked to ensure that unresolved activities are also followed through to conclusion. To facilitate a product- or department-specific evaluation, it may be expedient from an economic point of view to assign the additional or subsequent costs incurred to the deviations.
The results of the statistical failure category evaluation should be regularly available to the head of production, head of quality control and quality assurance on a regular basis so that appropriate measures may be initiated in response to undesirable developments, e.g. intensification of personnel training measures, bringing forward scheduled equipment maintenance dates, shortening maintenance or calibration intervals or galenical rechecking of formulations.
When using a database system, it should be remembered that GMP-critical data is managed within it which places demands on the validation status of the system.
11.K.7 SOP "deviations" - (example)
From the viewpoint of Good Manufacturing Practice, the handling of deviations is an extremely sensitive company issue, a uniform approach must be written down in a SOP in every case. An example is provided below
|
|
|
|
|





Summary The handling of deviations directly reflects the company's own concept of perception of quality in production: organisation forms are company-specific. Attention must be paid to deviations, research carried out on the causes and measures documented in the failure investigation report. |
