Risk management and quality management system
|
This section addresses the following questions:
|
Risk management acts as a very useful supplement to any quality management system (QM system) in two respects. First, risk management can be used to specifically optimise an existing QM system through the application of suitable methods for evaluating the system and regulations, and deriving appropriate consequences. Second, a QM system is also an ideal vehicle for implementing a risk-based approach across the board (at least for QM processes). This section explains both aspects in more detail. First it should be made clear, however, which prerequisites are required for which improvements to be achieved. The following explanations assume that a superordinate QM system is in place, ideally across the whole company, and that the included regulations are binding for all functions named within the scope (see chapter 1 Quality Management).
Through the implementation of the risk-based approach in the QM system, it is possible to achieve considerable improvements in terms of:
- Transparency of regulations and the relevant backgrounds,
- Acceptance of the system
- Achievement of 100 % internal compliance by defining appropriate requirements in the regulations
- Efficiency of the QM system through prioritisation of the topics and reducing the scope of regulations to the necessary level
Furthermore, asking appropriate questions can reveal gaps in the system.
1 Application to the QM system
A QM system consists of binding regulations that describe the internal implementation of external GMP requirements. In addition to formal compliance, the quality of products and processes is a clear objective. External specifications can frequently be interpreted to a greater or lower extent to allow for adaptation to suit the specific conditions within the company. Depending on the author of the internal regulation, this interpretation can be handled in different ways - with the result that internal regulations can become highly specific or too detailed, while others are so general that no extra level of company-specific precision is achieved in comparison with the external regulations.
An initial general but well-documented risk evaluation of the individual blocks of a QM system (e.g. validation, documentation, deviations, etc.) can help to make the process more objective. The initial risk evaluation is performed on a general level in order to achieve a good overview of the whole system relatively quickly and with a reasonable initial expenditure. In a second step, a detailed evaluation can then follow, which is subsequently reconciled with the general evaluation (see figure 1).
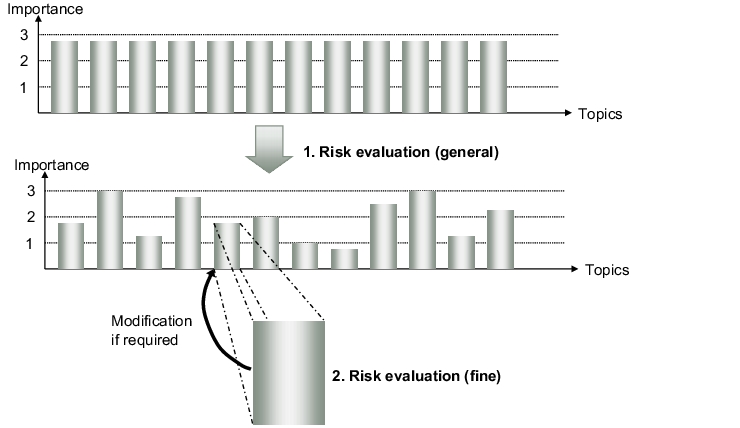 |
1.1 Evaluation of the system (rough evaluation)
You need a team, defined evaluation criteria, and a definition of the procedure for deriving conclusions.
The team
should be not too large, in order to avoid endless discussions. In general, the team should consist of experts in the area under evaluation (production, development, quality control) and a representative from quality assurance. Ideally this should be formed as a core team, in order to guarantee a similar level across the whole system (across all regulations). This core team can be enhanced by individually recruited experts from the specific subject areas (e.g. the QP responsible for releases).
Evaluation criteria
are also essential for ensuring comparability between the different subject areas. For a rough risk evaluation, risks relating to patients (quality, safety), compliance (external and internal), and cost-effectiveness should be assessed at a higher level. A classification, e.g. on a scale from 1-5, can be recorded together with corresponding explanations, and all criteria can be added to make a single total that is viewed as a key figure (figure 2). If necessary, the criteria can be assigned different weighting factors. For purposes of traceability and transparency, it is useful to include an appropriate and detailed rationale for the allocated grade.
|
Topic |
Patient risk |
External |
Internal |
Economic risk |
Global |
Total |
|---|---|---|---|---|---|---|
|
Topic 1 |
1 |
1 |
3 |
2 |
n |
8 |
|
Topic 2 |
3 |
5 |
4 |
4 |
y |
19 |
|
Topic 3 |
1 |
1 |
1 |
1 |
n |
5 |
In this example, Topic 1 is a local QM process (e.g. validation). The influence on the patient is very indirect, if validation, in a more specific sense, is only considered as the proof that a process is under control. There are no new realisations or specifications in the regulatory environment. Audit findings have shown, however, that deficiencies still exist in this area that must be addressed. An economic risk results from compliance deficiencies which, in the case of an inspection, may lead to sanctions, albeit not severe ones.
Topic 2 is graded higher - in this case there is a lot of movement in the external environment that must be addressed. Furthermore, this is also a global process, which means that a higher level of detail and/or the description of the process as a flow chart is applied in order to guarantee smooth cooperation between individual locations.
Topic 3 is viewed as non-critical (e.g. training). Well-established and functional systems exist at a local level, and thus the economic risk is also very low.
As mentioned above, the numbers in the table should be accompanied by explanations in the form of key words to guarantee traceability of the grading (for example, as comments in Excel).
Procedure for deriving conclusions
At the end of the process, a range of key figures is available for all topics. System-related conclusions, e.g. prioritisation for the revision, or revision intervals, can be derived directly. Further consequences, which are viewed more at the level of the regulation, can be determined from the answers to predefined questions. Examples: Level of detail of the regulations, description of "What" or "How", necessity for further, more detailed risk evaluations at operational level.
1.2 Evaluation of individual topics (fine evaluation)
The fine evaluation of the individual topics is performed as a part of the regular revision. Also in this case, the author should form a competent team of experts that is not too large.
The best approach is to start from the key words defined in the rough evaluation and discuss these in detail. The procedure in this case can be an informal risk analysis (see chapter 10.D.3 Informal method), or alternatively you can apply an FMEA (see chapter 10.F Failure Mode Effects Analysis (FMEA)). A combination of both methods can often be effective.
Simply asking "why" has proven to be a very simple but effective method. In doing so, you can read the existing regulation point by point, and question why the point is written in that way. In most cases, this is a surprisingly quick process.
It is essential to record the answers to the questions in writing, for three reasons:
1. It is often not clear what you really mean until you write it down.
2. It is a quicker method of coming to an agreement with all persons involved in the evaluation.
3. The result of the discussion is transparent for others who are not involved.
It often becomes apparent during the course of the discussions where which risks lie, and at which points which factors should or must be changed, abbreviated, or described in more detail, and how. This should be recorded (i.e. documented in key words) at this stage for future processing or revision of a topic.
Once the risks have been identified, they can be quantified (if practical).
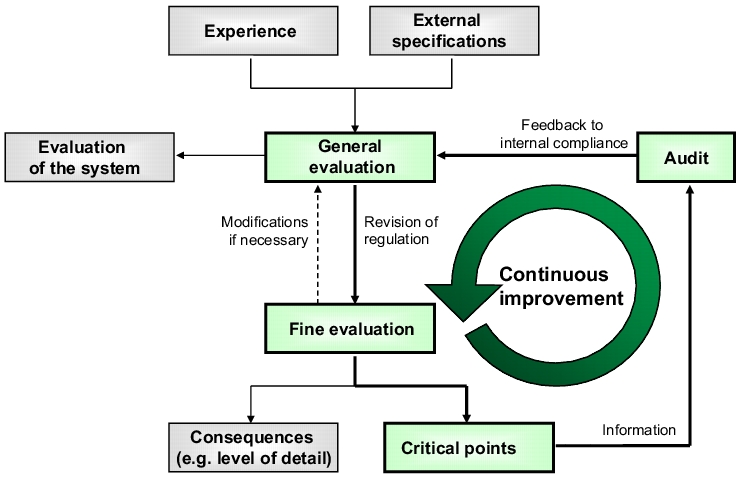 |
In the same way as for system evaluation, you can now derive consequences, or the results of the discussion can be documented separately. As a result, you end up with a list of critical points for this topic, which should firstly be reconciled with the general evaluation, and secondly also provide important feedback for the internal audit in order to draw specific attention to the issue. However, if you also ensure that someone from auditing is a member of the team for the general and/or fine evaluation, the continuous improvement cycle becomes a closed loop (see figure 3).
2 Implementation of risk management
via the QM system
Among the consequences resulting from the risk evaluation of the individual regulations, you can establish at which points a detailed risk evaluation is required at local and operational level. Using process validation as an example, we can see that the implementation can be represented by a 3-level model.
- 1stlevel: "what" specified by QM regulation - i.e. that critical parameters/processing steps must be defined and validated on the basis of a risk evaluation.
- 2nd level: implementation in a local SOP (local QM system), in which the "how" is defined (procedure, responsibilities, forms, etc.)
- 3rd level: application of the local SOP to a specific manufacturing process.
If this is implemented across the board throughout the whole system, and the advantages (objectivity through same evaluation criteria, transparency and traceability by answering the "why" question, safety due to evaluation in a team, etc.) thus become apparent, over time a risk-oriented mindset and outlook is automatically established, which is then applied to other processes and other questions.
2.1 Example of process validation
1st level
QM regulation (quote from an internal company guideline on process validation)
"[...] A risk evaluation is carried out in order to determine the critical steps/parameters that may influence the quality of the drug substance/medicinal product. The basis of this risk evaluation may be formed from the following:
- Scientific observations and particular results (e.g. deviations)
- Documented experiences from development and/or production.
[...] "
2nd level: local SOP
A local SOP contains specifications on how to define critical parameters. Using the manufacturing of drug substances as an example, this may be as follows:
- First step: determination of the critical synthesis steps using the HACCP method (see chapter 10.G Application example of a combined FTA and FMEA Combined FTA and FMEA (example)):
In this example (see figure 4), there are two critical control points (CCP) that require particular monitoring.
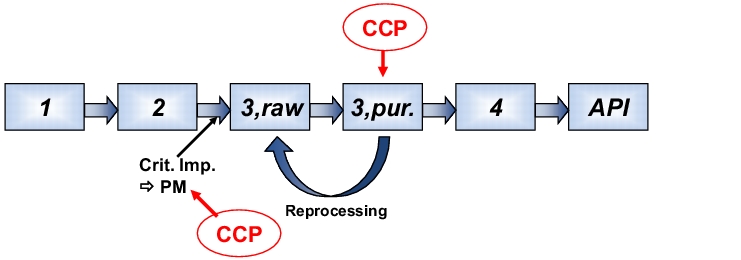 |
The first is process monitoring, in order to avoid the formation of critical impurities (because they cannot be reduced at the stage of the API). The second is the quality of the intermediate stage 3, which must always be guaranteed, including in an emergency by repeated purification, eg. reprocessing. In general, the final purification to the active pharmaceutical ingredient (API) is also validated, simply because any effect will have a direct influence on the quality of the active pharmaceutical ingredient.
- Second step: definition of the critical parameters for the individual levels. This can be viewed simply as a follow-on step to the HACCP in which you only refer to the identified critical levels or to the overall synthesis as a supplement to the HACCP. The method is a modified FMEA (see chapter 10.F Failure Mode Effects Analysis (FMEA) which follows a thorough preliminary analysis (see figure 5).
Figure 5 Differentiation of synthesis steps (IP = impurity)
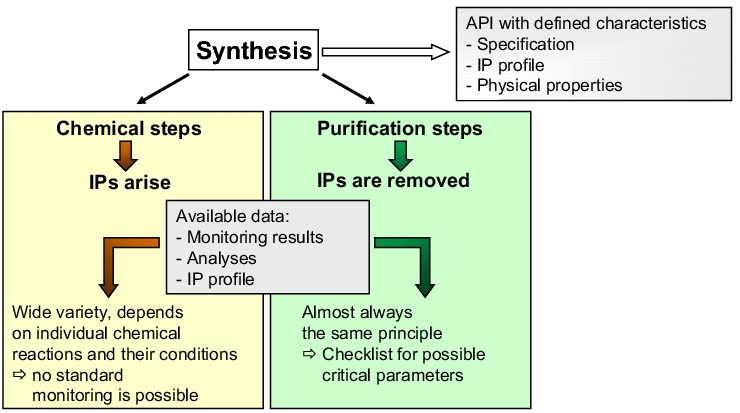
While evaluation of the chemical steps must be performed on an individual basis, purification steps (e.g. crystallisation) can be evaluated according to a uniform procedure (see figure 6).
|
Parameter |
Relevant |
Critical level |
Permitted |
References, |
|---|---|---|---|---|
|
Material |
||||
|
Quantity of solvent |
||||
|
Solvent:substance ratio |
||||
|
Solvent mixture |
||||
|
Quantity of excipients |
||||
|
Apparatus |
||||
|
Material |
||||
|
... |
||||
Here, the potential parameters that can have an influence on the success of the purification are listed systematically. The first step is a yes/no decision which sometimes automatically results from the process (e.g. no entry is made for Quantity of excipients, if none are used). The critical level describes the influence on the purification effect. If permitted limits exist, e.g. from laboratory investigations, these can also be entered. For practical reasons, notes should include a reference to the source from which the result was obtained (e.g. number in the laboratory notebook). In this way, it is possible to generate a document that is simple, but which essentially contains complete information on this purification step.
3rd level: application to a specific manufacturing process
The local SOP is implemented, the synthesis is evaluated in terms of its critical steps, and the steps are evaluated in terms of their critical parameters. The most important information on the synthesis is then described in the corresponding documentation. This provides a basis for the validation protocol.
|
Summary A QM system and risk management excellently compliment each other in two ways. First, risk management can be used to specifically optimise an existing QM system through the application of suitable methods for evaluating the system and regulations and deriving appropriate consequences. Second, a QM system is also an ideal vehicle for implementing a risk-based approach across the board (for QM processes and other processes). This can be clearly demonstrated using the example of process validation. |
