8.I Maintenance of the validated status
|
Here you will find answers to the following questions:
|
According to Section 7.3.8 General of the PIC/S guideline (PIC/S PI006), "Control of change to validated cleaning procedures is required. Re-validation should be considered under the following circumstances:
- Re-validation in cases of changes to equipment, products or processes,
- Periodic re-validation at defined intervals."
According to section 7.3.9, manual cleaning procedures must be checked more frequently (shorter time intervals) than CIP systems.
The supplementary GMP Guideline for qualification and validation (Annex 15) contains the following provisions for revalidation in paragraph 45:
"Facilities, systems, equipment and processes, including cleaning, should be periodically evaluated to confirm that they remain valid. Where no significant changes have been made to the validated status, a review with evidence that facilities, systems, equipment and processes meet the prescribed requirements fulfils the need for revalidation".
According to this, the validation cannot be considered as a linear process that is concluded as soon as the validation report is approved, but has to be interpreted as a cyclic process requiring ongoing monitoring (figure 8.I-1).
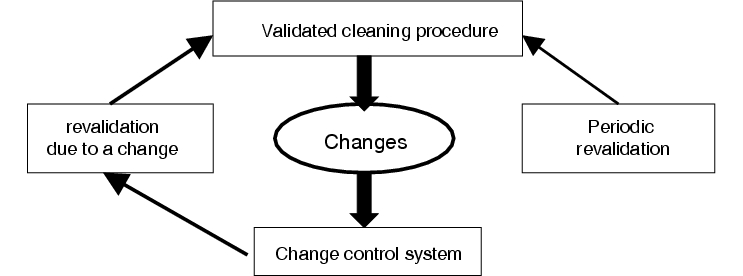 |
8.I.1 Changes and deviations
Each validated process is characterised by a number of parameters that affect the soundness of the validation if they are changed. A differentiation must be made between planned (i.e. controlled) changes, and spontaneous (i.e. uncontrolled) changes: the former are normally referred to as changes, and the latter as deviations (see chapter 19.C Change control).
A schematic representation of the various process steps for changes and deviations is provided in figure 8.I-2.
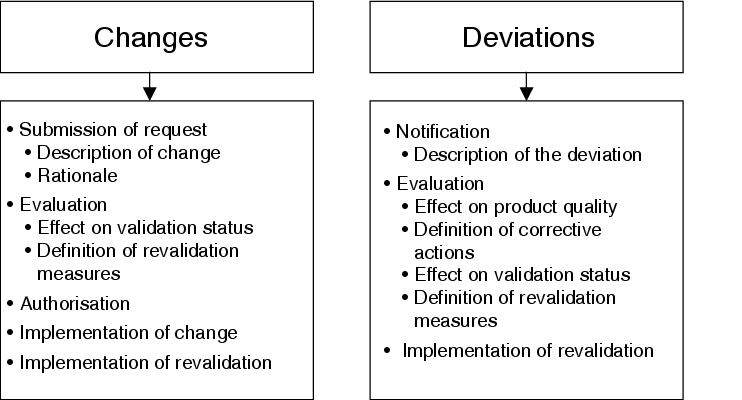 |
8.I.2 Change control
The soundness of the cleaning validation may be impaired by a number of changes. In principle the following cases have to be differentiated:
- Changes to the cleaning procedure
- Changes to the production equipment
- Changes to the product
In order to be able to assess the influence of changes on the validation status and initiate appropriate follow-up actions, change control procedures must be used to record and assess specific changes.
A prerequisite for an efficient change control procedure is to establish a connection between the aspects of the relevant change and the cleaning validation. The first process step is the change request; the second is the change assessment with definition of all necessary follow-up actions.
A number of difficulties may be encountered when linking the elements of a change with the cleaning validation during the first process step. It frequently happens that changes are requested by persons carrying out tasks that are not connected with the cleaning validation, and who are therefore not in a position to identify possible effects on the validation status.
Although requests can be submitted by a large and diverse group of persons, change requests are generally processed by a central functional unit within quality assurance. The awareness of this group to examine changes critically with view to possible impact on the status of cleaning validation has to be raised by appropriate training. In case of changes relevant to the validation, the persons responsible for change control must forward information to the persons responsible for cleaning validation. The latter must then decide on the type and scope of any revalidation measures required and inform the persons responsible for change control accordingly, as follow-up actions must be defined as part of the approval procedure for change requests.
The above demonstrates that close cooperation between the cleaning validation and change control functional areas is very important. To ensure a uniform process without interruptions the formal handling should, as far as possible, be detailed in writing as a standard operating procedure.
8.I.3 Revalidation
8.I.3.1 Change-related revalidation
The decision as to whether a revalidation must be carried out as the result of a change will always be made in each case, according to the circumstances. Similarly, the type and scope of the revalidation also depend on the particular situation. The following considerations should therefore be regarded as guidance only.
Changes to the cleaning procedure
As the cleaning instruction is the main focus of the cleaning validation, content changes (i.e. changes to the cleaning process itself) generally give rise to the need for a revalidation. Examples of these kinds of changes are:
- Use of a new cleansing agent
- Change in concentration of the cleansing agent applied
- Use of new cleaning utensils (sponges, brushes, etc.)
- Change of process parameters (pressure, temperature, time)
- Change in volumes of cleaning and rinsing fluids
Excluded are editorial changes to the cleaning instruction such as structuring, organisation, or layout, that do not affect the cleaning process itself. Revalidation is not required as a result of these kinds of changes.
Changes to the production equipment
Although changes to the production equipment can be many and varied, there are essentially two types:
- Changes to the CIP equipment
- Changes to/replacement of equipment parts
Examples of changes to CIP equipment are:
- Replacement of nozzle heads
- Changes to the dosing system for cleansing agent or its replacement
- Changes to pipe system (pipe length and diameter, water pressure)
No examples of changes to the equipment itself are given here as these are potentially too diverse.
When assessing an equipment-specific change, the following must be considered to determine the necessity and extent of a revalidation:
- Does the change affect the efficiency of the cleaning procedure?
- Are new product contact materials used?
- Are new critical locations appearing?
- Is the product contact surface changing?
The efficiency of the cleaning procedure may be adversely affected either by changes to CIP equipment or by changes or replacement of equipment parts. In this latter case it is especially due to the appearance of new critical points and the use of new product contact materials that the efficiency of the cleaning procedure is adversely affected (see below). A visual check of the production equipment provides an initial reference point to assess this situation. However, the only way to ensure compliance with established limits is through the analytical determination of residues.
New product contact materials may cause an increased risk of residues, if critical substances adhere to these more readily. This may be due to a higher surface rawness or the type of material used. For example, lipophilic active pharmaceutical ingredients, which can easily be removed from stainless steel or glass surfaces, may tend to form residues on plastic surfaces. It should also be taken into account that new product contact materials might have different recovery rates for the determination of residues.
New critical locations may appear if new parts are added to a production equipment or if existing elements are replaced by new ones having different constructions.
For changes to the production equipment attention must not only be paid to the emergence of new critical points, checks must also be carried out to determine whether the product contact surface has changed. This may affect the calculation of the limit in the sample, which reduces as the product contact surface of the manufacturing equipment increases, where the limit in the subsequent product remains constant. If, following an equipment-specific change, the resulting limit in the sample is lower than the value used as the basis for the validation, this new limit must be used for all subsequent validations. If the limit calculation is carried out as described in chapter 8.E Acceptance criteria and limit calculation, this measure applies for all equipment used to manufacture the critical product and not just for the production equipment where the change occurred. Furthermore, a critical review of all existing data must be carried out. If the results obtained to date are well below the new limit, it may be assumed that the recently calculated limit may also be complied with once the change has been implemented. If the results obtained to date are close to or exceed the newly calculated limit, optimisation of the cleaning procedure for the relevant production equipment should be considered followed by revalidation.
Changes to the product
The following product changes may affect the cleaning validation:
- Changes to composition
- Changes to manufacturing process or production equipment
Changes to composition are always possible when products are reformulated. In most cases, these are changes to one or several excipients whereby technological issues such as compressibility, disintegration or content uniformity may be decisive for the reformulation. Although in these instances the physico-chemical and pharmacological properties of the active pharmaceutical ingredient that are the main focus of the risk analysis do not change, the behaviour of the overall formulation may change during cleaning which means that the suitability of the established cleaning process must be checked in every case.
Changes to the manufacturing procedure may affect individual manufacturing steps, or even the entire manufacturing operation. A possible scenario is, for example, the transfer of a product compression from a tablet press type "x" to another - maybe newly aquired - tablet press type "y". It is also conceivable that the manufacturing operation could be partially or entirely changed as a result of reformulation. An example of this type of situation would be a changeover from wet granulation to direct compression, or vice versa. Changes to the manufacturing procedure inevitably imply the need for recalculation of the product contact surface and limit in the sample. The resulting follow-up actions have been explained previously in the section entitled "Changes to production equipment". The likelihood of a significant limit change is naturally greater when one or several items of equipment used within the manufacturing process are replaced as opposed to the aforementioned replacement of equipment parts.
If the manufacturing operation is changed, a revalidation must be carried out for all additional new equipment.
The remarks in this section apply primarily for critical products - i.e. products that have been graded as critical on the basis of a risk analysis due to potential cross-contamination of active pharmaceutical ingredients. However, it should also be considered that products previously regarded as non-critical may become relevant to the cleaning validation as a result of changes in their composition or changes to the manufacturing process, but such instance is not be regarded as a revalidation in the true sense of the word.
8.I.3.2 Periodic revalidation
Both the PIC/S guideline PI006 and the Annex 15 to the GMP Guideline require periodic revalidation according to specified time intervals to check and ensure the validated status.
As this requirement is very non-specific in nature, it leaves a large number of questions open.
- How long is this time interval allowed to be?
- Does the complete validation (e.g. for several critical substances on one type of equipment) have to be repeated or is a reduced scope of validation sufficient (e.g. for one critical substance only)?
- Are three positive test runs required again or will one positive test run be sufficient?
- Are all sampling locations to be tested again, or is it possible to reduce this scope?
There are no general answers to these questions that reflect the present state of the art. Many pharmaceutical manufacturers are currently in the completion phase of the original validation program, whereby at the same time new validation projects are emerging from the introduction of new products and equipment. In the light of this situation, a revalidation concept must be developed that not only satisfies official requirements for continuous review and maintenance of the validated status but is also feasible regarding time and costs.
In conjunction with a revalidation concept, the following aspects must be defined at the outset:
- Time interval
- Linking with change control procedures
- Type of test implementation
When determining the time interval, it should be considered that according to the PIC/S directive (PIC/S PI006), manual cleaning processes should be checked at shorter time intervals than automated cleaning processes.
Periodic revalidation can be streamlined by linking this with change control procedures. If, for example, the specified time interval is 2 years, cleaning procedures that have not been revalidated as a result of changes in this 2 year period may be prioritised over those that have. For the latter, the time interval for the periodic revalidation can be redefined starting from the time the change-related revalidation was carried out. This makes it possible to prevent change-related revalidations to cleaning procedures followed by periodic revalidations with only short time intervals in between.
The type of test implementation in straightforward cases may consist purely of a document review, and in less favourable cases, repetition of the entire validation study. Although no further remarks are provided by the PIC/S guideline (PIC/S PI006), Annex 15 to the GMP Guideline states:
"Where no significant changes have been made to the validated status, a review with evidence that facilities, systems, equipment and processes meet the prescribed requirements fulfils the need for revalidation."
The term evidence used here allows room for interpretation. In the most favourable case, this clause implies that a pure document review can take the place of an experimental revalidation provided that it is clear from the documents that no changes relevant to validation have been made. However, it is also conceivable that authorities will take the view that tests must be carried out (determination of residues in this case) in order to provide evidence. As Annex 15 first came into force in June 2001 and no specific reference is made to cleaning validation in paragraph 45 titled Revalidation, it remains to be seen which interpretation will be used in practice.
In every case the basis of a periodic revalidation should be a document review using a defined questionnaire. Some guiding principles for this are provided in figure 8.I-3.
|
Questionnaire for document review |
|---|
|
Review of cleaning instruction
|
|
Review of qualification documentation
|
|
Comparison of the current product assignment with the product assignment at the time of validation
|
|
Comparison of the current manufacturing instructions with the manufacturing instructions at the time of validation
|
|
Review of change control documentation
|
8.I.4 New products and equipment
Where a new concept for a cleaning validation programme is developed, the risk analysis for the bracketing and validation matrix, which is in turn derived from this, is based on the current product range and available production equipment.
If new products and equipment are added later on, this raises the question as to whether a cleaning validation is necessary. The aim here should be to integrate new products and equipment logically into the existing concept to ensure its consistency.
8.I.4.1 New products
When carrying out bracketing, distinct active pharmaceutical ingredients are identified as critical because, in physico-chemical and pharmacological terms, they represent the worst case for cleaning validation.
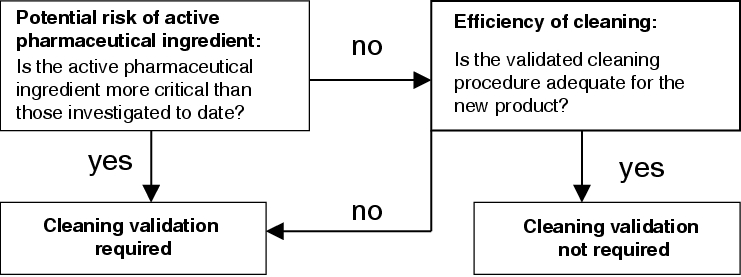
If new products or active pharmaceutical ingredients are added, their potential risk must be compared with that of the critical active pharmaceutical ingredients already identified. To assess the potential risk, it has to be checked first to which of the risk groups the new active pharmaceutical ingredient should be assigned. Within the relevant risk group the solubility, therapeutic dose and fraction in the formulation are compared with the data for the critical active pharmaceutical ingredients already identified (for procedure, see chapter 8.D Establishing the scope of validation). A cleaning validation must be carried out for the new active pharmaceutical ingredient if it is less soluble, if it has a lower therapeutic dose, and/or if its fraction in the formulation is higher than the critical active pharmaceutical ingredients already identified.
If all three parameters are less critical than those of the critical active pharmaceutical ingredients already identified, a check should be carried out in the next step of the risk analysis to verify whether the established cleaning procedure may be used for the new product: if this is the case, a cleaning validation is not necessary.
However, if the new product requires a new or modified cleaning procedure, validation of this procedure becomes mandatory and a cleaning validation for the new product must be carried out.
A graphic representation of the risk analysis procedure for new products is shown in figure 8.I-4.
 |
8.I.4.2 New equipment
If new production equipment is introduced at the company, a decision should be made during the qualification phase about the necessity and scope of the cleaning validation. The following issues should be addressed:
Type of equipment
If the production equipment represents a completely new type, a cleaning procedure must be developed and validated in accordance with the intended range of products.
If the same type of production equipment is already available, additional questions must be addressed to determine the necessity and scope of the cleaning validation.
Cleaning procedure
If the product range intended to be manufactured using the new production equipment includes products for which a cleaning procedure other than the established one applies, this procedure must be validated. In so doing, the validation must be carried out for the most critical product to be cleaned using this procedure.
If all products to be manufactured on the new production equipment can be cleaned using the cleaning procedure that is already in place, the validation status must be checked in the next step.
Validation status
If a cleaning validation has already not yet taken place, this should be carried out taking the new equipment into account.
If a cleaning validation has already taken place, the product assignment for the new equipment must be compared with the existing validation scope established to date in the next step.
Product assignment
A new worst case cleaning validation must be carried out if products graded as more critical than products for which a cleaning validation has already been carried out are to be manufactured on the new equipment. The risk assessment for the products must be carried out in accordance with the information provided in chapter 8.I.4.1 New products.
If products graded as less critical than products for which a cleaning validation has already been carried out are to be manufactured on the new equipment, no further cleaning validation is required.
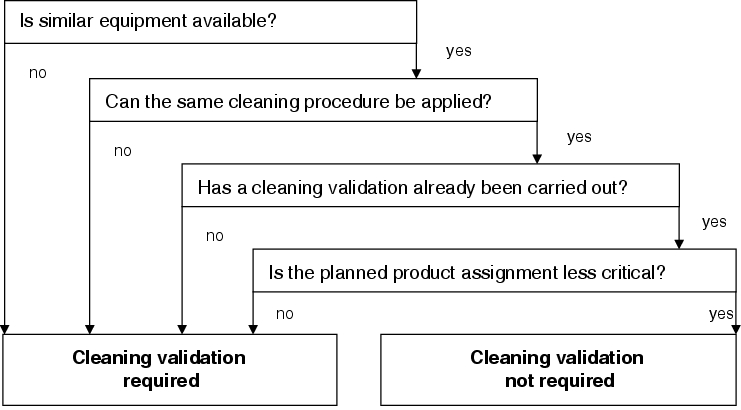
This procedure is shown as a graph in figure 8.I-5, and may be summarised as follows:
A cleaning validation for new production equipment is necessary
- if no similar equipment is used at the company, OR
- if a different cleaning process to the one already used for similar equipment is required for products to be manufactured on the new equipment, OR
- if the products to be manufactured on the new equipment can be cleaned using an existing cleaning procedure for similar equipment that has not yet been validated, OR
- if products to be manufactured on the new equipment are more critical than products for which a cleaning validation has already been carried out using similar equipment.
A cleaning validation for new production equipment is NOT necessary
- if similar equipment is used at the company, AND
- if the products to be manufactured on the new equipment can be cleaned using an existing cleaning procedure for similar equipment, AND
- if this cleaning procedure has already been validated, AND
- if products to be manufactured on the new equipment are less critical than products for which the cleaning validation has been carried out.
 |
8.I.5 Deviations
While changes are planned in advance with the general aim of making improvements, deviations occur spontaneously - often due to defects or deficiencies.
Concerning validation two types of deviations are generally to be distinguished:
- Deviations from the validation protocol
- Deviations from the acceptance criteria
For example, the following deviations from the validation protocol may occur:
- Samples have not been taken at all sampling locations.
- Samples have been taken at the wrong sampling locations.
- Mistakes have been made during sampling (material for swab test, type of solvent, volume of rinsing solution, ...).
- Validation has not been carried out for the prescribed product.
All of these deviations are not process relevant as they only relate to the implementation of the validation. In general, these deviations should be noted at the latest by the time the validation report is compiled and can be rectified by repeating the faulty test.
When deviations from the acceptance criteria occur, established limits are exceeded. This may take the form of single or multiple events. A deviation that occurs once may be caused by a random error that is not necessarily process-relevant, whereas deviations occurring several times are likely to be caused by systemic errors or defective processes. The essential problem in assessing an exceeded limit is that it is only possible to differentiate between single and multiple events with a reasonable degree of certainty once all data from the validation study are available - yet at the same time, appropriate measures should be taken once the deviation has occurred. The fact that only three test runs must be carried out for a critical product makes it even more difficult to differentiate between random (or non process relevant) and systemic (or process relevant) errors. It follows that if a limit is exceeded once at a random sampling location, this can easily be declared as a random error whereas if a limit is exceeded several times at the same sampling location, a systemic error may be assumed. What happens, on the other hand, if the limits for three test runs at three different locations are exceeded?
The causes of exceeding limits and the subsequent corrective actions are varied and dependent in each case on the type of cleaning procedure used, the production equipment being investigated and the general conditions of the validation as regards personnel and organisation. However, some basic procedures for the processing of deviations still apply.
Initially the deviation is documented appropriately and forwarded to the person responsible for cleaning validation, who then performs a risk assessment with regard to potential contamination of the subsequent product and initiates immediate measures (if required) to ensure the safety of the product. Limit exceedence has a wide range of consequences as illustrated by the following examples:
Example A:
A facility for the preparation and dispensing of a solution is sampled by means of a rinse test. The rinse test is carried out across the entire product contact surface. The concentration of the sample solution shows that the acceptable residue limit in the facility has been exceeded. As the entire manufacturing operation is carried out on this facility, the limit in the subsequent product will also be exceeded. The product manufactured on the facility following the cleaning validation must be rejected as the acceptable quantity of residue of the previous product will be exceeded.
Example B:
Sampling is performed on a high shear mixer by means of a swab test at five critical locations. The result of one sample is above the acceptable limit for the sample whereas the results of the other four samples are well below it. The risk assessment, for example, could read as follows:
"The acceptable limit for the sample was exceeded at one of five critical locations. The proven residue quantity at the remaining critical locations is well below the acceptable limit. Therefore it may be assumed that residues on the remaining non-critical surface of the equipment are also well below the acceptable limit. It follows that the acceptable residue quantity in the facility is not achieved or exceeded."
In contrast to example A, the high shear mixer is only used for one step within the manufacturing process and the product contact surface is therefore only part of the entire product contact surface for the manufacturing operation. This aspect can also be dealt with in the risk assessment roughly as follows:
"In addition to the high shear mixer, other production equipment is used to manufacture the critical product. No exceedences of limits or acceptable residue quantities were observed in those facilities. Therefore a contamination of the subsequent product beyond the acceptable level can be certainly excluded ."
This kind of risk assessment may be supported by calculations. In this case it will prove to be positive if non-critical locations in the production equipment are also sampled. If sample results for critical and non-critical locations exist, an extrapolation may be made with reference to the product contact surface of the production equipment. An example of this is shown in figure 8.I-6.
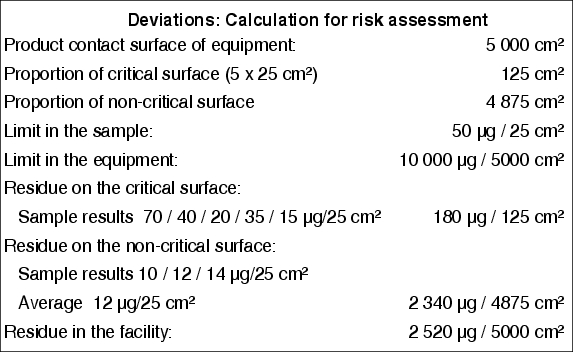 |
Example B demonstrates that a single limit exceedence does not necessarily represent a problem in terms of the quality or safety of the subsequent product. Nevertheless, limit exceedences must be investigated as they may be indicative of cleaning procedure deficiencies or implementation errors.
Whereas the risk assessment should be carried out directly after a limit has been exceeded, it may prove expedient to determine the cause of a limit exceedence not earlier than after completion of the validation, as only then all data will be available and differentiation between single events and multiple events will be possible.
Figure 8.I-7 shows schematically the possible causes of a limit exceedence.
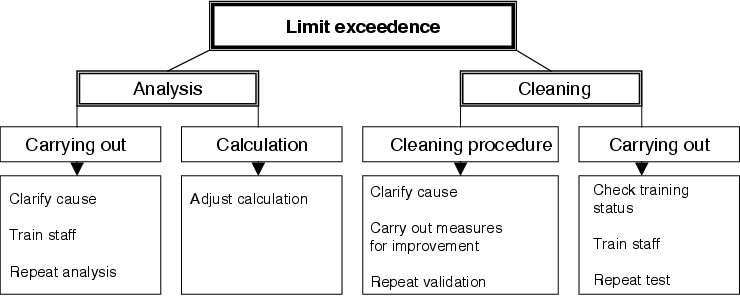 |
The first step that should be taken once a limit has been exceeded is to check whether the error can be traced back to the analytics. The error may be a calculation error or an implementation error. An error in the calculation may be easily rectified by carrying out the calculation again correctly, whereas a suspected implementation error must be confirmed by a replicate analysis. In so doing, a problem that often occurs in practice is that the sample solution required to perform the replicate analysis is no longer available. If there is a suspicion that the limit exceedence could be traced back to an analytical error and if it is not possible to perform a replicate analysis, the only available option is to repeat the validation test.
If deficiencies in analytics can be excluded as the reason for the limit exceedence, the cleaning procedure must be investigated for possible causes of failure. In doing so, a general differentiation must be made between deficiencies in the cleaning procedure itself and implementation errors. This means that an unsatisfactory cleaning result due to incorrect or careless implementation of an optimised cleaning procedure, is just as likely as an unsatisfactory cleaning result due to an inadequate but correctly implemented cleaning procedure.
The first step therefore is to check whether the cleaning has been carried out correctly. Such a check might be based on existing records, or consist of interviews with the staff authorised to carry out the cleaning. An error made during implementation of the procedure is not process relevant, and it is therefore sufficient to repeat the defective test run. Appropriate retraining must be given beforehand and retrained staff should be involved in the repetition of test runs.
If no implementation error is found, the efficiency of the cleaning procedure must be examined. In this case it is definitely appropriate to wait until all the results of the entire validation study are available: for example, limit exceedences may occur at only one specific location in the equipment, or at several different locations. If the exceedence occurs only at one location, it may be possible to make improvements by intensifying the mechanical cleaning effort at the location concerned. If the exceedence occurs at several locations, a possible solution is to optimise basic criteria - e.g. higher concentration of cleansing agent or longer application time. If the cleaning procedure has to be reworked due to limit exceedence(s), the entire validation must be repeated.
|
Summary: Validated cleaning processes must be subject to change control. Further, revalidation is necessary in order to maintain the validated status where a differentiation must be made between change-related and periodic revalidation. An efficient change control procedure must ensure that the effect of changes on the cleaning validation is identified, and that any necessary follow-up action is initiated. Checks must be carried out to verify whether revalidation is necessary following changes to the cleaning procedure, production equipment and products. A suitable time interval must be determined for the periodic revalidation. Implementation may be streamlined by linking this to the change control procedure and by reducing the scope of the validation based on a review of data and documents. The validation requirements for new products and equipment may be determined by carrying out a risk assessment with the aim of integrating the new products and equipment into the existing validation concept. When carrying out the validation, deviations from the validation protocol and deviations from the acceptance criteria may occur. If a limit is exceeded, a risk assessment must be carried out promptly for the subsequent product. On the other hand, root cause analysis and definition of corrective and follow-up actions often makes sense only after completion of the validation study when all available data may be taken into account. |
