Here you will find answers to the following questions:
|
If you consider the actual implementation of a pharmaceutical quality management system, then it becomes apparent that the systems available in the company are mainly based on the nine chapters listed in the EU GMP Guideline. These mainly focus on the steps and processes that have a direct relationship with the manufacturing and quality control of the respective products. There are also other systems that are effective in the periphery and ensure the quality of the products. Figure 1.C-1 shows the subjects covered in the guideline.
Chapter 1 Quality assurance system Chapter 2 Personnel Chapter 3 Premises and equipment Chapter 4 Documentation Chapter 5 Production Chapter 6 Quality control Chapter 7 Contract manufacture and analysis Chapter 8 Complaints and product recall Chapter 9 Self-inspection |
Other specific subjects are described in appendices 1 to 19 of the EU GMP Guideline (see chapter C EU GMP Guide), such as
- sterile drug products,
- biological drug products,
- clinical trial samples,
- drug products made from blood or blood products,
- manufacturing of active pharmaceutical ingredients (API).
However, these do not make any significant new requirements (other than the chapter on APIs) of the quality management system itself. Only basic requirements for manufacturing and quality control are stated in more detail and specified or explained in greater detail for particular processes. The requirements must, of course, be included in the available systems, but simply require the tightening of individual measures or more detailed implementations.
The following chapters explain some of the contents listed in more detail and give detailed descriptions of their implementation in the company's quality management system. If subjects are dealt with in detail at other places in the GMP Manual, cross references are given.
Almost all individual systems listed below are linked with each other and only operate on their own in the rarest of cases, if at all. Where applicable and important, the connection to other systems is mentioned and the crucial links are cited and explained in more detail.
1.C.1 Management responsibility
The EU GMP Guideline requires the existence of a quality assurance system. This system must ensure that the product quality requirements listed are implemented and complied with. However, the quality assurance system cannot exist and be further developed alone, but rather it requires active support in the company. Verbal support alone is not sufficient. Instead, specific responsibilities must be accepted for the system, for its functionality and for its further development. In accordance with the EU GMP Guideline, these responsibilities should be shared by the management board, the staff in the company and the suppliers and sales companies.
The question now is which employees are given which responsibilities and to what extent are they relieved from the management board responsibilities mentioned in the guideline. And what role should be attributed to the Quality Unit?
1.C.1.1 Responsibility of key personnel
In order to be able to specify the activities together with the responsibility of the aforementioned employees in the company, chapter 2 of the EU GMP Guideline stipulates that the employees who have to actively support the quality management system are primarily key personnel.
Such key personnel include the head of production, the head of quality control and a Qualified Person. Included in their range of tasks, they are responsible for ensuring that the implemented quality management system is used in an appropriate manner.
1.C.1.2 Responsibility of the management board
In order to keep the entire system up-to-date and effective in the company and to steer all work in the right direction, there is a need for overall responsibility. This overall responsibility can, however, only be assumed by a person or group of persons who have extensive authority to make decisions and can also determine the direction in a company. The legislator intended the management board for this purpose, it is the highest management level for any company. For a company floated on the stock market, it should be a member of the board of directors, for a limited company it should be a member of the management board. Only these people can determine the direction and level of implementation, as they have the necessary authority to make decisions and also have access to the company's finances.
Any company that has concerned itself with the establishment, implementation and, in particular, maintenance of an expedient quality management system, knows that this system causes not insignificant costs during implementation. Even during ongoing maintenance and necessary further development, increasing amounts of money and resources are required. Unfortunately these costs cannot always be estimated, nor can the effectiveness of the quality management system be calculated.
Hereinafter, the term management board is used as a synonym for the board of directors, the management or other persons in a company, who have authority to make decisions and budget.
Since the responsibility for the overall quality management system implemented in the company lies with the management board, the following questions must be asked:
- Which specific responsibilities must stay within the management board and cannot be delegated?
- How can the management board exercise its responsibilities?
- How can the management board provide proof of its responsibilities?
The answers to the questions raised here are given below, which help to involve the managing directors responsibly in the system and demonstrate acceptance of responsibility.
The EU GMP Guideline does not provide any further information or help on the above-mentioned questions. It therefore makes sense to look for help in related standards and guidelines and derive corresponding implementations for the management board from these. The points listed below are taken from DIN EN ISO 9001/9004:2000 and, if considered in detail, also meet the intention of the EU GMP Guideline. The internationally recognised management standard can be referred to for support, especially as the direction of the DIN EN ISO standard is being increasingly pursued in the future development of the implementation and monitoring of the quality management system.
Figure 1.C-2 briefly lists the expectations of the management board, which are expressed in more detail in the following chapters. However, each company is free to consult other or additional points regarding the management board's responsibility and formulate this accordingly.
Management board responsibilities |
Brief description of implementation |
|---|---|
Definition of a quality policy |
The management board should compile a general policy defining the implementation of a quality management system and aligning the system with the general objectives of the company. |
Definition of quality objectives |
Targets, which must be checked and adjusted each year, must be defined for all employees in a management position (head of department and higher). They must be in agreement with the quality policy. |
Support of the QM system |
The key points here are meetings with the head of the Quality Unit, receipt of different documents that prove the effectiveness of the quality management system and compilation of the management review. |
Establishment of an organisation |
Compilation and signing of an organisation chart for the company; set up Quality Unit as staff function and define reporting channels |
Delegation of partial responsibilities and activities in job descriptions |
The company should have job descriptions which are to be counter-signed by the management board from the head of department level at least. |
Discussion and decision on resources |
Together with the definition and follow-up of quality objectives, discussion and decision-making regarding resources can be managed in meetings with the Quality Unit. |
Implementation of a quality assurance/management department |
Establishment and implementation of a department or person who has a supporting and supervisory function for the quality management system |
Management review |
At least once a year, the managing director should review the quality management system. |
The ISO standard may not be a direct requirement for the pharmaceutical industry, but the DIN EN ISO is globally accepted as a management standard.
The FDA has moved one step closer to EN ISO 9001:2000 in a Draft Guideline (Guidance for Industry, Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations of September 2004). This specifies the ISO standard as a structure and requirements specification for the management level.
It is important to prove that the requirements made of the management board are actually realised and implemented in their daily work. A purely verbal commitment to the individual points is not sufficient. Rather, there should be a sufficient quantity of documents providing proof of compliance with the established responsibilities. They should show when, how and to what extent the management board has dealt with the individual requirements and which quality management decisions it has taken.
1.C.1.3 Definition of quality policy
First of all, the management board should establish a quality policy for the company. In addition to the general company policy, this should also include points which primarily deal with the quality of the products as well as the quality of daily work. Furthermore, the policy must also give information on how and to what extent the quality management system should be supported by the overall company management, and also the extent to which it should be further developed. The quality policy should contain generally valid statements, from which further, specific obligations for the company can be derived for the system.
Quality policy points could be:
- Information on personnel:
- Resources
- Education
- Professional training
- Implementation of a Quality Unit function/department
- Obligation towards the customer (authorities are also customers):
- Meeting the customers' requirements
- Achieving customer satisfaction
- Conformity between registration and manufacturing
- Observation of guidelines:
- Observation of and compliance with the bodies of rules and regulations - primarily the GMP rules
- Checking and possibly adjusting the system when the rules are modified
- Continuous monitoring of the quality management system:
- Implementation of a review system in the form of self-inspections
- Involvement of the suppliers and service providers in the system, specific requirements, monitoring
- Carrying out expedient and controlled changes
- Qualified and validated processes:
- Maintaining the qualified and validated status - particularly after changes
- Qualifying and validating new processes
The quality policy is to be formulated and signed by the management board. All employees can be notified of the policy either via the Intranet or by hanging it up in the form of a written document, which can be part of a quality management handbook or the Site Master File. By signing and publishing the quality policy, the management board shows all employees in the company that quality management is an important and advisable factor in the overall company workflow and is desired, supported and developed by the top management level.
1.C.1.4 Definition of quality objectives
Such an agreement should be completed with the heads of department and the division managers. From a GMP perspective, these established objectives should primarily contain points regarding the quality of the processes and thus of the products. Likewise, the scope in which the QM system will be supported, and with which specific projects, should also be recorded.
While the quality policy represents a formulation which, once established, does not experience any short-term changes, the quality objectives must nonetheless be agreed at least once a year with the superior or management board.
They are to be recorded in writing and can contain the following points for a quality unit, for example:
- Number and time of execution of audits
- Number and time of execution of training courses
- Compilation and review of SOPs
- Review of the validity of procedures
- Training on new/changed rules
- Determination of room for improvement in a specific process,
e.g.: complaints about a particular product
The quality objectives for the Quality Unit thus only comprise a systematic goal. These are points that are necessary and pertinent for the maintenance and continuation of the implemented quality management system.
It is important for quality objectives to contain specific information on the content and time of fulfilment of the tasks together with a specific objective, if feasible. The last example listed for the Quality Unit is intended as a target value for determining the room for improvement for complaints. Specific objectives could be the processing time, costs or reduction of frequency of complaints. The quality objectives are to be signed by the management board and the respective head of the unit.
1.C.1.5 Support of the quality management system
In order to effectively support a quality management system, general and specific information on the system must be obtained at regular intervals. The most effective method has proven to be a regular meeting together with the head of the Quality Unit. In such a meeting, the management board finds out about specific issues in the company which concern the fulfilment of the EU GMP Guideline. In order to plan the meeting effectively, a specific agenda should be compiled so that both sides can prepare themselves. For example, the points listed in figure 1.C-3 can be used as an established agenda.
|
Points for discussion with the management board |
|---|
|
Finally, a record should be compiled containing specific actions either for the Quality Unit alone or also for the management board. This record will show the extent to which the management board has met its obligations from the EU GMP Guideline, and with which points for decisions.
1.C.1.6 Deciding on resources
When the management board holds discussions about resources, the main objective is often personnel capacity. In principle, this also applies for the management board's responsibility in respect of the quality management system. It should be possible to show that the personnel situation in all areas and departments is adequate to carry out the necessary activities. The term resources, however, not only covers the number of persons in the personnel cover, but also their qualification and training (resources of qualified staff). Staff training can be defined and proven in the form of an annual training plan - similar to an annual audit programme. SOP training courses cannot always be included in the plan if they cannot be planned in advance. Training for SOPs must be carried out depending on the situation and must be individually defined during compilation and/or change before becoming applicable (see also chapter 1.C.7 Training).
In addition, the resources to be provided by a company also include the available premises and equipment. These should offer sufficient space and capacity for performance of the necessary, quality-related steps. For the equipment, for example, it should be shown that the equipment and facilities used are continuously serviced and maintain the qualified status.
Not least of all, the resources also include external companies who work under manufacturing contracts. The qualification of external contractors is of particular importance, especially if they have to carry out quality-related steps. The management board must therefore define measures and describe procedures for how contractors or service providers are qualified and remain qualified (see chapter 1.C.10 Qualification of suppliers and service providers).
The management board is responsible for ensuring sufficient availability and qualification of all listed resources. The company itself will decide on the definition of "sufficient". This should either be described in an SOP or in the quality management handbook. The Quality Unit cannot solve resource problems by itself, but can discuss these points either in the audit reports or in general during the meeting with the management board.
1.C.1.7 Management review
The management board must carry out and prove its responsibility in specific points. The lip service provided in the past and viewed as adequate cannot persist, in particular in light of future development. The FDA's guideline (Guidance for Industry, Quality System Approach to Pharmaceutical Current Good Manufacturing Practice Regulations, Draft Guidance, September 2004), sets clear expectations of the management board in terms of the implementation of and responsibility for the quality management system, which will also have an impact on the European area. Therefore, regular meetings with the Quality Unit and a written assessment of the quality management system are advisable, preferably at the end of the year. The management review may contain points in line with DIN EN ISO 9001/9004:2000 (see figure 1.C-4). Depending on the company, additional topics that are deemed to be necessary may be added to these points.
|
Content of a management review |
|---|
|
However, compilation of the review alone is not enough. It should be sent to all employees in a management role so that the corresponding points can be incorporated in objective agreements (see chapter 1.C.1.4 Definition of quality objectives). In addition, points for the optimisation of the quality management system can be gathered in the respective areas. Implementation can be checked by the Quality Unit during the self-inspection.
|
Summary The management level is responsible for establishing the quality management system based on the information in the EU GMP Guideline. Monitoring of the functionality and further development of the system can then be reviewed and managed through regular meetings with the Quality Unit. |
1.C.2 Change management system
|
Here you will find answers to the following questions:
|
1.C.2.1 Definition of terms
During the manufacturing or quality control of a pharmaceutical product, problems can occur in many places until the release and sale of the product. Increasingly it is the case that deviations are observed during manufacturing, changes are requested for the process or specifications are not complied with in quality controls. For all these eventualities, the Quality Unit must define a procedure that allows for concentrated and targeted processing of these problems.
The problem areas list can generally be summarised under one term, "change management", as they usually concern changes in the broadest sense of the word. This term was chosen on the one hand because these three systems are repeatedly asked for during an inspection and, on the other hand, to give a superordinate term that enables uniform understanding. It is also possible to go one step further, at least for deviations and changes, and process them in one system using a form. In the initial stages of the processing in the OOS system, it is possible to proceed according to the general form for deviations/changes. Thereafter, however, due to the specific requirements from the laboratory area, a separate form is required.
Thus, the following subsystems can be defined under the term "change management":
- Changes
- Deviations
- OOS
Definition: changes
This includes all changes that are planned and known in advance. This means targeted and desired changes to sequences, processes and, in many cases, also to the products themselves, which have some influence on quality. This includes changes in the following systems, for example:
- Documents
- SOPs
- Specifications
- Equipment
- Personnel
- Laboratory standards
- Software
- Rooms
- Suppliers
The most frequent changes are made in documents and especially in SOPs, which do not have to be managed by a formal change procedure in Europe. The FDA, however, expects this for this system too.
Definition: deviations
Deviations are observed whenever an unplanned move away from a prescribed sequence or an established standard occurs.
In contrast to a change, the problem with a deviation is that it is only detected once it has occurred. So, for example, the production of a product has already progressed and the problem cannot be monitored and controlled in advance as is possible with a change. Deviations become extremely problematic when they are detected in the batch record review to be carried out at the end of production or in the final analysis. Deviations occur frequently, for example, in or if
- Procedure parameters,
- Documentation of the work carried out,
- Checking the environmental conditions,
- Checking/monitoring the equipment and measuring devices,
- Requirements from the SOP or other instructions are not followed,
- Manufacturing parameters are outside the range in terms of their established specification,
- Manufacturing conditions (e.g. hygiene conditions) are retrospectively discovered to be non-compliant with the specifications,
- Organisation/personnel.
Definition: OOS
All results that are outside the specification according to analysis of the final product in the quality control are called OOS results.
|
Definition: OOS (out of specification) |
|---|
|
"The term OOS results includes all suspect results that fall outside the specifications or acceptance criteria established in new drug applications, official compendia, or by the manufacturer." from FDA Draft Guidance for Industry: Investigating Out of Specification (OOS) Test Results for Pharmaceutical Production (chapter D.9 Guidance for Industry: Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production) |
1.C.2.2 Processing of changes and deviations
An almost identical system can be implemented for deviations and for changes. A change is only brought out of the procedure for deviations at a few specific points. The remaining points can be considered as identical for deviations and changes.
In most cases, it is necessary for a team to carry out further processing following receipt of the change/deviation. In rare cases, the deviation/change can be completed purely through interaction between the specialist department and the Quality Unit. The formation of this team means all necessary aspects and queries from all areas can be discussed and decided on.
The members of this team should include at least the following people:
- Head of manufacturing
- Head of Quality Control/Qualified Person
- Head of marketing authorisation/registration
- Head of quality assurance
If necessary, additional competent employees can be included. Such additional members often include the head of engineering or the head of research and development. However, it is not absolutely necessary for the team members to be recruited solely from among the heads of the respective areas. In the operational departments in particular, it can be advisable to involve other competent employees. Participation of the head of marketing authorisation/registration is absolutely necessary as a change/deviation and its further processing could affect the marketing authorisation and thus the marketability of the product.
The team's task is to assess the documented change/deviation primarily in respect of the following criteria:
- Influence on the quality of the directly affected product
- Influence on other products
- Influence on the validated status
- Influence on marketing authorisation/registration
The change/deviation is assessed by means of a risk analysis. If a risk analysis is already available, it can be used for the discussion. If there are not critical implications to the change/deviation, it is processed in collaboration between quality assurance and the respective specialist department, the measures are documented and it can be completed by quality assurance.
If one or more of the above criteria are fulfilled, it could be that immediate measures are required in a further step, in particular for deviations. These immediate measures must be arranged by the responsible persons (see figure 1.C-6).
|
Immediate measures |
Responsible |
|---|---|
|
Rejection of the batch |
Qualified Person |
|
Rejection of the manufacturing processes |
Head of manufacturing |
|
Rejection of equipment, facilities or rooms |
Head of the specialist department in question |
|
Making contact with the authorities |
Head of marketing authorisation |
|
Other measures |
Head of quality assurance |
The next step is to determine and define the further procedure via a risk analysis. The Quality Unit should lead the discussion as team leader and arrange for the immediate measures established. The work from the risk analysis is to be coordinated and followed up by the Quality Unit. Processing of changes/deviations can be completed when all measures established in the minutes of the meeting and in the risk analysis have been processed and implemented. This is the task of the Quality Unit. The legally responsible person agrees to the measures taken. The basis for decision is often the discussion in the team.
The deviation system in particular is very closely linked with the Corrective and Preventive Action system (CAPA), see also chapter 1.C.4 Corrective and Preventive Actions (CAPA). During processing, remedial action is first defined, which should help initially control the problem. In further processing, the preventive actions are defined, which should prevent repeated occurrence of the deviation.
The points formulated on the form or the documents attached to the deviation are to be considered as immediate measures (corrective action). They do not have to be listed again in the Corrective and Preventive Action system. A cross reference is sufficient in this case. The established remedial actions are to be copied into the CAPA system and followed up there.
In connection with the processing of changes, it is necessary to point out a problem that occurs very often in companies and is only detected at a later time. In addition to the planned and known changes, other changes can occur that are not directly recognised as such. Yet these become obvious in the further course of manufacturing and/or quality control through problems in processing (detected here as deviations) and must be dealt with retrospectively.
Examples include:
- Repair and maintenance work:
- Installation of a new part (wear and tear)
- Importing of a new software version
- Exchanging identical parts
- Changes in packaging material
- Changes in raw material
- Supply with energy (gas, water)
- Staff changes
The system to be established by the Quality Unit must ultimately be able to record all changes - and thus also these indirect changes - in the company and have them processed centrally. For more information, see chapter 19.C Change control and chapter 11.K Deviations.
1.C.2.3 Processing of OOS results
There is no direct guideline in European legislation regarding the processing of OOS results. Monitoring by the authorities is based on the Draft Guideline published by the FDA in 1998 as a general basis (see chapter D.9 Guidance for Industry: Investigating Out-of-Specification (OOS) Test Results for Pharmaceutical Production). In accordance with this guideline, OOS results only had to be processed for the product analyses which were applied when checking the finished products. However, it is now also customary to use the procedure for analysis results that occur during production, that is, during the in-process control..
|
|
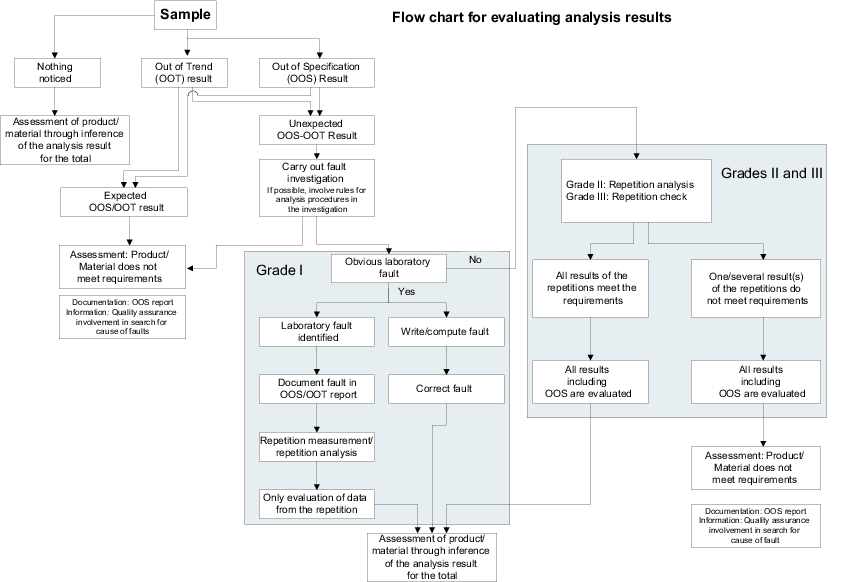
The method can also be used for stability data, but should be limited to the inspection of laboratory data. As the processing of OOS results involves a truly extensive and complex procedure, it is not recommended to extend it to "OOS" results in engineering. The Quality Unit should establish other procedures for this purpose
Strictly speaking, OOS can be considered as a special kind of deviation, with the limitation that it is only observed in the laboratory and here, in particular, during analysis. It is therefore advisable to have the corresponding SOP compiled by the head of quality control as a specialist and approved by the Quality Unit. Figure 1.C-7 shows an example of an OOS procedure. Further information is provided in chapter 14.H Out-of-specification results.
1.C.2.4 Involvement of external companies
The involvement of external companies in the internal change management system must be very intensively regulated. There is a risk that the external service providers will not detect changes, deviations or OOS results as critical events, because, for example, the regulatory background is not known. On the other hand, the multitude of possible changes, deviations or OOS results from all external companies associated with a company must be contained and kept to the necessary level. A quality management system carefully coordinated with the external companies provides for an effective filter in the information circuit. The following points have been determined as advisable measures:
- Audit of the company and comparison of the change management systems with the internal system
- Signing of a joint change management SOP
- Use of a joint form
- Definition of reporting criteria
- Regular training of employees
- Good qualification system for external companies
- Retain tried and tested companies
- Include conditions for a change management system in the contract
- Include key SOPs in the contract together with the version number
In order to be one hundred percent certain, an additional measure can be established as an active system. When sending a certificate, together with the required product or analysis values, a named person (head of the unit) should certify on the certificate or delivery note that no changes have been made to the procedure and no deviations (and thus also OOS results) have been ascertained.
|
Summary Changes and deviations are to be carefully processed and controlled in their execution so that the pharmaceutical quality is not impaired. In addition, it must be checked if the registration or marketing authorisation of the products is affected. Particular attention must be paid to external companies that are to be as deeply involved as possible in the company-internal system. |
1.C.3 Complaints and recall
|
Here you will find answers to the following questions:
|
The quality of the pharmaceutical products, which must be guaranteed in principle through the validity of the manufacturing procedure and a final quality control before dispatch, can never be 100% guaranteed. On the one hand, this is due to the fact that manufacturing always conceals a certain number of imponderabilities, which cannot even be excluded by extensive validation, in-process checks during production, and final inspection; On the other hand, it is due to the fact that only a quite specific sample volume of the sales product is subject to quality control before release and thus before sales. For these reasons, it may be the case that complaints from the market regarding the product reach the company.
1.C.3.1 Definition of terms
When dealing with defects in drug products, the EU GMP Guideline speaks of complaints. The defect may mean that the intended healing effect does not occur as expected, and in the worst case that it has no healing effect or causes other medical problems (see figure 1.C-8).
|
Examples of defects in drug products are: |
|---|
|
In addition to complaints, grievances must also be mentioned, which are received in the company and relate to the product in general and not directly to defects in the drug product itself. This includes, for example, problems with dispatch or communication. Upon first consideration, these do not have a direct influence on the product quality, but relate to the company's service provision.
Grievances received can thus include:
- Damage to packaging
- Expiry of the expiration date
- Unsatisfactory dispatch
- Excess/insufficient delivery
- Incorrect delivery
- Time lag in information/communication
- Poor information management, etc.
Complaint can be used as a collective term, and need not always refer to problems in the quality of the product. It is therefore important to distinguish between these two cases in the quality management system.
In addition to external complaints, internal complaints can also play a role. These occur whenever departments pass work or even products/intermediate products between each other and the quality of the work/product or the supporting documentation does not correspond to the expected requirements or regulations. They should also be taken into consideration when setting up a quality management system, as a product quality problem could ultimately also occur in these cases if they are not adequately and conclusively processed. In a well-structured quality management system, however, internal complaints are viewed as deviations and are processed in the corresponding system (see chapter 1.C.2.2 Processing of changes and deviations).
1.C.3.2 Processing of complaints
Complaints as described under grievances are not to be considered in detail here. They can, however, be processed using the same procedure (SOP, form) as quality problems. Some points from the SOP and the supporting form are not to be processed.
In principle, complaints with an influence on the product quality cannot be processed by the Quality Unit alone. Nearly all departments and areas are involved in the process, from purchasing to the warehouse and delivery. Only rarely can problems with the product quality be traced back directly to a specific point or sequence during root cause analysis and thus be unequivocally accounted for. In most cases, more intensive root cause analysis is necessary, which very often includes the reference sample - be it the finished product or the raw material.
1.C.3.3 Responsibilities
For processing reports that have become known about drug risks, the legislator has provided for a person, the drug safety manager, to collect the reports, assess them and arrange the necessary measures internally and externally. The drug safety manager works very closely together with the authorities. Based on the European directive, his responsibility extends only to the processing of side effects and is not to be dealt with further in the following observation. The Quality Unit can only offer support for the processing of side effects.
It is thus important to split the implemented quality management system into the three interrelated processes (see figure 1.C-9).
|
|

It is advisable to compile an SOP defining how legally prescribed activities are to be processed by the drug safety manager, and how all other complaints are to be processed by the Quality Unit. Collaboration between both positions is necessary and should be clearly and unambiguously described in the system.
1.C.3.4 Compilation of a standard operating procedure (SOP)
Depending on the size and scope of the company and the processing, several instructions may be necessary. The operations must be described starting with receipt of the complaint through to final processing, feedback to the customer or, in the most unlikely case, carrying out a recall.
Both processes can thus be jointly described in the instruction. Under responsibility, a note should be made that in the case of a legal complaint (side effect report), the drug safety manager, or otherwise the Quality Unit, are to be notified for further processing.
The SOP for complaints should primarily be compiled by the Quality Unit. To define the individual steps for the drug safety manager in accordance with legal requirements, the corresponding cross references should be incorporated into the drug safety manager's instruction in collaboration with him.
All processes to be carried out are to be described according to their processing sequence and the responsibilities and necessary activities in the different departments and areas must be addressed. The corresponding process form should reflect that the processes are listed chronologically and guide the processor in his activities.
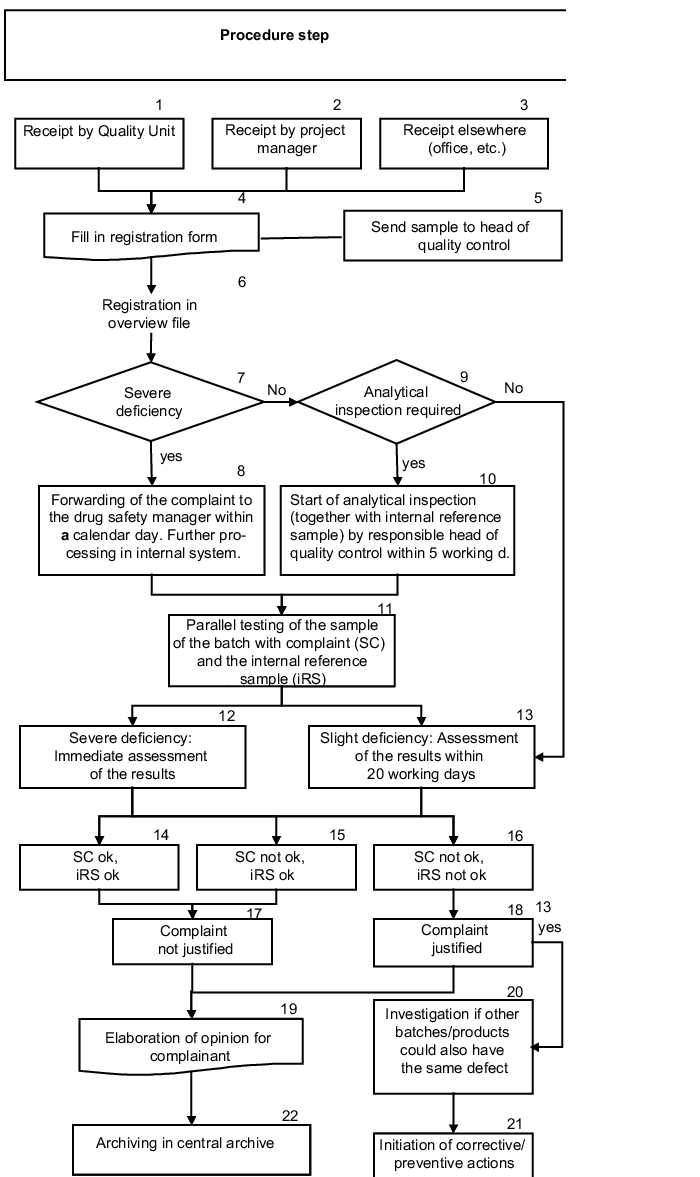
Figure 1.C-10 shows the complaints processing process.
|
|
|
|
1, 2, 3 all recipients |
The requirements of this SOP apply regardless of where the complaint is received. |
|
4, 5 all recipients |
Form; project manager and other: either fill in the form yourself and send it to the head of QA or call the head of QA. Head of QA: return call to complainant for the purpose of completion. Always arrange immediate transfer of a sample of the defective product to the head of quality control. |
|
6 QA |
Immediate entry in an overview file |
|
7, 8 Head of Quality Control |
Severe deficiency: the head of quality control arranges the start of the analytical testing for comparison with the internal reference sample within one calendar day. |
|
9, 10 Head of Quality Control |
Slight deficiency: the head of quality control decides if analytical testing for comparison with the internal reference sample is necessary and, if relevant, arranges this within five working days. |
|
11 Head of Quality Control |
The analytical testing is carried out under the supervision and instruction of the head of quality control. |
|
12, 13 Head of Quality Control |
In the event of severe deficiencies, the head of quality control immediately assesses the results, or for slight deficiencies, he assesses the results within 20 working days of receipt of the complaint. Processing is carried out in accordance with the form. |
|
14-18 Head of Quality Control |
If SC and iRS are ok, or if the iRS is ok and the SC is not ok, the complaint is not justified. If both samples are not ok, the complaint is justified. |
|
19 Head of qUality Control |
Following submission of the results and their assessment, the head of quality control compiles the opinion for the complainant. |
|
21, 21 Head of Quality Control |
Complaint is graded as justified: in collaboration with the head of QA, the head of quality control checks if other batches of the same product or of other products also have the same deficiency. If this is the case, both jointly initiate suitable remedial action (possibly via the QA team). |
|
22 Head of QA |
The QA is responsible for archiving the overall documentation for at least ten years following completion of the procedure. |
Receipt of the complaint
In order to ensure a controlled process from the outset and to obtain exactly the information required for further processing, the corresponding questions should already be available in the documents via specific querying routines when the complaints are received.
The form for recording the complaint should be available to employees at all positions in the company whose activity lead them to have contact with external customers. In this way, they can ask for the necessary information directly when a complaint is received. Such employees include the marketing and sales department, in addition to the product owner (product manager). Both are the first to have contact with a customer in most cases. However, it should not be forgotten that the form is also to be saved in the production, quality control and Quality Unit areas, especially for the management staff. In order to ensure standardised use of this form, regular training is required for all staff who record and further process complaints.
In any case, it is necessary to provide the sales representative/pharmaceutical consultant with the form and include them in the training. He is able to properly record the complaint directly on site during a visit or call with the customer and to ask for and enter all information required for internal processing. The form filled in by him is then forwarded directly to the Quality Unit. In accordance with the written regulation, this initiates further internal processing.
Tasks of the Quality Unit
The Quality Unit has a key function in the overall process. It receives the complaint after it has been recorded and carries out an initial assessment. In many cases, completion is possible here. However, criteria which enable completion by the Quality Unit at this stage must be established in the quality management system. If further processing and clarification of the problem is necessary, then the Quality Unit can obtain the help of other departments and experts at any time (through team building too). It obtains corresponding information and work from the individual experts.
It is important that the Quality Unit - or at least quality control - hear about when product complaints are received. In the system, it must be ensured that a direct link to the corresponding complaint is created. A query routine for returned products should be included in the form.
In the event of a drug product risk, QU forwards the procedure directly to the drug safety manager who, in accordance with the legal provisions, carries out further processing in his special system.
Technically, complaints are a deviation (see chapter 1.C.2.2 Processing of changes and deviations). The identified problem should therefore be analysed to see if it also has implications in other batches of the same product. If equipment, methods or premises are involved, there could be general problems in the procedure processes. In this case, other products, which are not directly connected with the processed problem, might be affected.
When all results from the areas and departments are available, the Quality Unit assesses the complaint and decides on further measures alone or in the committee. After the Quality Unit gives its opinion based on the available data, the complaint can be concluded in accordance with established conditions. It is advisable and also necessary to involve the legally responsible people, such as the head of manufacturing, the head of quality control and the Qualified Person, in the decision-making, at least for the completion of the complaint and the definition of possible further measures.
The result of the processing of a complaint with corresponding reasoning is forwarded to the customer and also provided to the sales department and pharmaceuticals consultant. The system can also send the processing results to the customer via the sales department or the pharmaceuticals consultant.
Upon completion of the internal processing of the complaint in the individual specialist areas, the entire procedure with all appendices is returned to the Quality Unit, which is responsible for completing the overall procedure and for providing information to the customer or internal position.
The Quality Unit can also archive the entire procedure. In many cases, these documents are also sent to the product owner. In the SOP, the Quality Unit must define which position carries out archiving.
1.C.3.5 Recall
It may be the case that products about which complaints are received have to be recalled from the market. The company must implement a system for this purpose, which deals in detail with the process of a recall and defines exact requirements for the individual steps.
With recalls, it is often the case that companies soon experience a hectic rush and people start making their own decisions. It is therefore all the more important to define a clear reaction rule for all employees and to have it prescribed as binding by the management board. In this case, it is best to employ the drug safety manager in a coordinating role, as the authorities must be informed.
The following points are to be established in a corresponding SOP:
- Who initiates the recall (can be the Quality Unit together with the head of quality control)?
- How is the customer informed?
- How is the product retrieved?
- How is the recalled material handled?
- Checking for completeness of the recalled material
- Communication with the authorities (via Quality Unit or drug safety manager)
- Definition of time frame
In the warehouse, there must be a closed and separate room for the recalled products so that mixing with approved products which are ready for dispatch cannot occur. Specific labelling of returned, incoming products is also very important. They must be clearly labelled as such and the label must be found on every packaging unit.
Thankfully, recalls are not carried out very often, so this crisis situation only very rarely occurs in a company. However, this presents a problem for a functioning quality management system because all processes have to intertwine smoothly when a recall has to be activated. To deal with this particular problem, a recall can be simulated from time to time during a self-inspection using documents to simulate the product. The process should be viewed in the same way as a fire drill. It is hoped that the situation will never occur, but it is best to practice in case it does.
An important note to add here is that the Quality Unit must establish an effective system for dealing with the returned products internally. In this context, please refer to the Reworking and Reprocessing systems, which are closely connected with complaints with product recall (see chapter 11.L Reworking). In most cases, however, it is advisable to completely destroy the returned product, as further processing does not appear expedient from an economical perspective.
When processing complaints, the necessary consequences must be established in the form of corrective and preventive actions (CAPA) (see chapter 1.C.4 Corrective and Preventive Actions (CAPA)).
1.C.3.6 Trend analysis
The Quality Unit, together with the drug safety manager or in separate groups, can have the task of compiling and processing the data received for a trend analysis. The following parameters can be included in an analysis for this purpose:
- Processing period for a complaint, starting with receipt by the Quality Unit and ending with the assessment
- Capacities used in the individual departments/areas
- Product costs if replacement was delivered
- Amount of complained product
- Other affected batches, if relevant
- of the same product class
- of other product classes
The trend analysis data can then be included in the management review (see chapter 1.C.1.7 Management review).
|
Summary Complaints and grievances must be processed accurately and in accordance with detailed descriptions. This must be a key element of a properly functioning quality management system, as the quality of the products and provision of services to the customer are also involved. Its external impact must not be underestimated. |
1.C.4 Corrective and Preventive Actions (CAPA)
|
Here you will find answers to the following questions:
|
There are repeatedly cases in which the processes for manufacturing and quality control as well as for the system do not run as was actually intended. Corrective intervention must then be taken to get back on the tracks specified by the corresponding SOPs and bring the quality management system back into equilibrium.
So, for example, corrective actions which in most cases are followed by preventive actions, must primarily be carried out when processing
- Deviations
- Out of Specification (OOS)
- Complaints
- Deviation report of an inspection
- Trend analysis
Corrective actions must be taken first and then preventive actions. This system is called a CAPA system (Corrective Actions and Preventive Actions). Generally speaking, the CAPA system must always be used when problems occur with the processes or the product.
Originally, the CAPA system was not defined in the EU GMP Guideline. However, if you consider the processes, for example, of the processing of deviations, then it becomes clear that the CAPA system has always been in use, even if it was not known as such. CAPA as a system element stems from the field of medical devices regulations together with EN ISO 9001 and the FDA CFR 820 regulation. However, it is increasingly being used as a separate management element in the pharmaceutical industry.
1.C.4.1 Definitions
|
Corrective action |
A corrective action is an action taken to clear the detected problem directly. |
|
Preventive action |
This is an action taken and suitable for avoiding recurrence of a detected fault or problem in future, or preventing it with greater certainty. |
The definition in accordance with DIN EN 9000:2000 deviates from this (see figure 1.C-12). It is important to be aware of these differences during the discussion with certified companies.
|
3.6.5 Corrective action Action taken to eliminate the cause of a detected fault (3.6.2) or another undesired situation
|
|
3.6.6 Correction Action for eliminating an identified fault (3.6.2)
|
|
3.6.4 Preventive action Action taken to eliminate the cause of a possible fault (3.6.2) or another undesired possible situation
|
1.C.4.2 Quality management system for CAPA
In a pharmaceutical company, there are two main different types of CAPA systems that can be implemented.
Area-internal CAPA system
For this purpose, a separate SOP that defines and identifies links to the other, above-mentioned systems (deviations, complaints, etc.), is compiled in the areas or departments. Starting with problem processing, the operation is transferred to the new, internal system and the link is documented with a separate ID.
The advantage of this system is that a clear separation between the two processes can be documented. Each system can be presented and discussed in its own right.
The disadvantage is that these are two separate processes which must contain a cross reference in order to be able to link the two processes. This means that information can be lost or incorrectly transferred. Only both documents together are legible and comprehensible for the CAPA system.
Company-wide CAPA system
This second case is an easier system for CAPA. At the end of the above-mentioned problem processing, a request to formulate, carry out and implement corresponding CAPA actions is made on the same documentation form. After problem processing and processing of the defined CAPA action, the Quality Unit can complete both procedures together, which are also associated with each other.
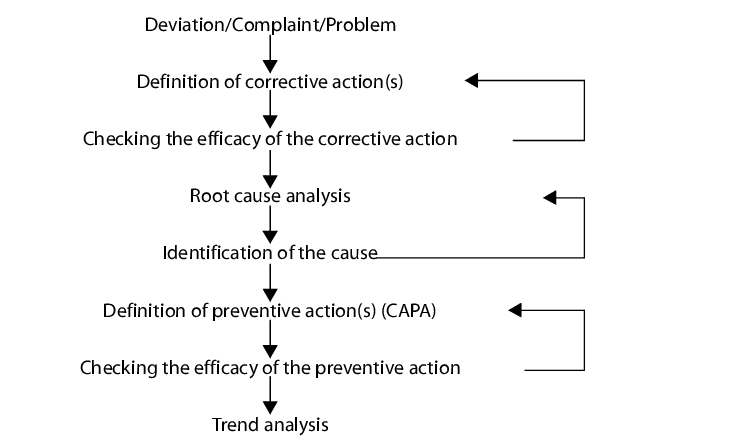
In order to understand the CAPA system correctly, it must be viewed in connection with the succession of different individual systems (problems). In its execution, it is at the end of a longer chain of quality management systems which extends from discovery of a fault or problem, to processing of this fault, through to definition of actions against recurrence and verification of their efficiency.
Contents of an SOP
|
|
In both of these possibilities, it is important to describe the CAPA system in an SOP, in which the processes are established together with the responsibilities and the necessary documentation.
The CAPA system can also be defined in the quality management handbook, together with the general description of the other quality management systems. In any case, after processing deviations, out of specification (OOS), complaints, deviation report of an inspection and trend analysis, it is advisable to urgently regulate the further processing of the problem in the CAPA system.
It must be recorded as a required point in processing; even if no specific item results as a further preventive action following exact analysis of a problem. In this way, however, there is documentation in all cases that thought was given to a further action in the meaning of CAPA.
As with processing of deviations or complaints, the CAPA measures can also be further processed and assessed in a committee, which is invoked and steered by the Quality Unit. The composition of this committee can be the same as for the original problem.
In order to simplify the selection of corresponding preventive actions for the decision-maker, a list for the processing of deviations, for example, can be added at the end of the SOP, listing the specified CAPA actions, such as:
- Staff training
- Changes to SOPs/documentation
- Changes in the process structure
- Increase of inspection cycles
- Discussion of resources
- Intensification of work
- etc.
In this connection, it should be pointed out that an increase in requirements is not always the only preventive action. There are also cases in which simplification of structures or documentation can bring improvement.
At the same time, measures should be defined to check the efficacy of the preventive action.
Figure 1.C-13 gives an overview of the processes of a CAPA system. In addition to linear processing of a fault, the meaningfulness and effectiveness of the actions taken should be queried repeatedly.
|
Summary The CAPA system is increasingly used in the processing of other systems, in order to complete the processes with expedient actions. In particular for deviations and complaints, this system has proven itself to be an expedient completion. |
1.C.5 Risk management
|
Here you will find answers to the following questions:
|
For a long time, risk management has been in use in many industries, such as in finance, insurance, health care or even generally by authorities in the monitoring of pharmaceutical companies. In recent years, the pharmaceutical industry has increasingly acknowledged risk management as an expedient and supplementary tool in its GMP-regulated processes. In discussions with the authorities, questions are increasingly raised about the critical steps in the manufacturing and control of a drug product and about mastery of these steps. In general, questions are more frequently being raised about potential risks in internal processes. These questions can only be answered properly with the help of a risk analysis.
The GMP rules do not directly mention the implementation of a risk analysis. However, it is repeatedly mentioned that the company is obligated to deal with the critical steps or critical parameters in a procedure to minimise or control them.
Risk management is used for
- Changes to procedures and systems,
- Deviations in procedures and systems,
- OOS results,
- Qualification of equipment, rooms and facilities,
- Validation of procedures,
- Complaint processing,
- Auditing,
- Selection/change of suppliers and service providers,
- Changing of starting materials.
Risk analyses are always to be carried out when
- critical or quality-determining parameters have an impact on the processes, systems or products,
- changes have been made to valid and established/mastered steps, or
- deviations have occurred.
1.C.5.1 Aims of risk management
Patient protection
This primary aim is clearly specified in the ICH Guideline Q9, Quality Risk Management (see chapter E.7 ICH Q8: Pharmaceutical Development), and should be the top priority in all pharmaceutical companies:
|
"The evaluation of the risk to quality should ultimately link back to the protection of the patient. "(ICH Q9) |
Procedure safety
Critical points of a manufacturing procedure or other processes can be elaborated starting in Research and Development, for and during clinical trials and lastly in manufacturing and control. As a result, operations and processes can be optimised and the available resources used in a reasonable manner. By implementing prospective risk observation, possible hazards can be averted in advance.
When processing defects (see chapter 1.C.3 Complaints and recall), the following questions can be asked:
- What influence does this or that operation, this or that result have on the further procedure for the product and thus for the patient?
- How can the identified risk be diminished or controlled?
- How can recurrence be prevented?
- How can the processing under discussion be implemented effectively?
Only through risk analysis can the problem be effectively processed and completed, in order to allow for corresponding follow-up (see chapter 1.C.4 Corrective and Preventive Actions (CAPA)).
Two not insignificant positive side effects should be mentioned in this context. By carrying out targeted risk analysis, the available resources can be used in a targeted and expedient manner when summing up the necessary work derived from this. The work is gradually carried out chronologically according to key points and the weighting of the task, rather than all at once or as and when possible. The resources can then be discussed efficiently with the management level according to urgency, and deployed accordingly.
In the case of monitoring by the authorities or for inspections of other companies, the focus has recently been increasingly placed on the critical steps of a process or the critical points of a product. The well documented risk analysis can be used as a starting document for this.
|
Summary A risk-based approach is recommended when dealing with changes, deviations, OOS results, qualification and validation, audits and supplier qualifications. This type of approach steers the focus directly to the critical aspects. The authorities, above all the FDA, also support the introduction of risk management. The ICH Q9 deals exclusively with this. |
1.C.6 Qualification and validation
|
Here you will find answers to the following questions:
|
Sequences and processes must be established that, with a high level of certainty, also guarantee products that correspond to the specifications and always reproducibly provide the same, required quality.
Comprehensive quality control on the final product could be used before sales of each batch to convince oneself that the specifications have been complied with. The procedure is not generally wrong, but conceals two major hazards.
1. You can never be certain that all quality related parameters have actually been adequately tested. This point carries all the more weight, considering that only a statistically secured quantity from the manufactured batch is tested during the final analysis. A statement must then be made about the entire sales product based on this random sampling.
2. If the fault (inadequate quality of the product) is detected at the end of the value-added chain, it is often not possible to carry out a correction, or only with disproportionately high expenses. From a business management perspective, this is the most unfavourable time to detect or even eliminate a fault.
In order to counteract the problem of late fault recognition, the authorities, and thus the pharmaceutical rules and regulations require that the manufacture, the quality control methods and all supporting systems be validated as far as necessary. This is based on a risk analysis that has previously been carried out. Only safe, robust, reproducible and thus valid manufacturing procedures continuously guarantee a consistently high product quality.
1.C.6.1 Tasks of the Quality Unit
The task of the Quality Unit is initially to define and establish the individual systems for qualification and validation. It is advantageous to specify the company-wide description and general structuring of these systems, together with the responsibilities, in the quality management handbook. This can be dealt with in a joint chapter together with the written description of the support provided by the company management. Based on this binding statement by the management board, an SOP is then compiled, either for qualification and validation together or individually. The decision on whether one or two SOPs are compiled at this stage depends on company policy. When defining the processes in two separate SOPs, it is important to demonstrate the complex connections between the qualification, the validation and ultimately the risk analysis.
The Quality Unit checks all other documents compiled for compliance with the requirements of this SOP and the regulatory and company-internal requirements. If they are logical and conclusive, the Quality Unit releases the documents. The Quality Unit is to watch out for any additional claims or changes in the documentation and must initiate their integration. Only then can the documents be released.
The proper processing of the specified tasks in respect of the individual stages of qualification are to be monitored by the Quality Unit. In the case of extensive qualifications or validations, the status of the work should be audited in the meantime either based on the previously received documents or on site during the work. This makes it easier to inspect and assess the documents at the end of the activities.
The release of rooms, buildings, equipment or facilities for validation after qualification can only be undertaken by the legally responsible person (head of manufacturing or head of quality control). The same applies for validation. The Quality Unit can only provide support in these cases and carry out a detailed review, which in many cases cannot be fulfilled by this group of people.
When compiling the final report, the Quality Unit should ensure that a corresponding release note appears on the document.
1.C.6.2 Tasks of the management board
Due to the responsibility of the management board for the quality management system and, in particular, for the registration-compliant manufacture of products, it has the task of establishing the qualification and validation system and monitoring its application. The management board should support the description of the systems, the responsibility, execution and updating of the qualification and validation in the quality management handbook and sign it as company policy/quality policy.
With this clear statement on the support of and need for this work to be carried out, the employees are more strongly obligated to implement it. In many cases, it is advisable to also have the qualification and validation protocols defined by the management board. There are two reasons for this. On the one hand, the management board recognises the costs behind qualification and validation, and on the other hand, resources and times are used for this work that must be offset from the normal work. In both cases, it is important to keep the management board informed.
1.C.6.3 Quality management system for qualification
The work described in chapter 1.C.6.4 Quality management system for validation, together with the differences listed here, result in a complete quality management system for carrying out qualification.
In order to make it easier for the employees to work through the individual qualification points, the Quality Unit can create a catalogue of questions, which must be worked through from DQ through to PQ (see figure 1.C-14, figure 1.C-15 and figure 1.C-16). When carrying out qualifications, it has repeatedly become apparent that a set of basic questions has to be processed, which can be specified in advance in the form. A significant advantage in this is that the qualifications can be compared with each other and the key points are not forgotten.
The form for carrying out the individual qualification levels, however, must have additional space for other important questions to prove the proper installation and function of the equipment or facility.
Prerequisites for qualification
Before qualification can be carried out, important basic prerequisites must be created, without which reasonable processing is not possible. When compiling a qualification protocol, these points are to be formulated as necessary requirements and should be inserted in a defined form for the protocol by the Quality Unit:
- The measuring devices used must be calibrated and have a sufficient level of precision for checking the unit of measurement.
- The risk analysis must have been completed and approved.
- The staff must be trained (if necessary).
- Valid SOPs must be available for carrying out the work, in particular for the technical monitoring and inspection of the equipment and facilities.
- The rooms and buildings must be qualified.
Checklist for installation qualification of equipment/facilities
|
Equipment |
|||||
|---|---|---|---|---|---|
|
Type |
Manufacturer |
Serial no.: |
|||
|
-Inv. no. |
Internal equipment no. |
||||
|
Site |
Operator |
||||
|
o New equipment since: |
o Equipment adopted since: |
o "Old equipment" |
|||
|
Action |
yes |
no |
not |
Date |
Comment |
|
Sketch/photo/diagram available |
|||||
|
Contractual service description available |
|||||
|
Technical documents (operating instructions, circuit diagrams, RI plans, etc.) available and transferred to Techn. Documentation |
|||||
|
External maintenance agreement available |
|||||
|
Connections (electricity, water, gas, or other energies described in the specification) properly executed |
|||||
|
Individual components agree with the components listed in the service description |
|||||
|
Alarms properly connected |
|||||
|
Site report made to technical service |
|||||
|
Person responsible for the equipment named |
|||||
|
Comments/deviations: |
|||||
|
Evaluation |
|||||
|
The IQ was carried out by: |
The IQ was assessed and released by: |
||||
|
Name/Date ... |
Name/Date ... |
||||
Checklist for operational qualification of equipment/facilities
|
Equipment |
|||||
|---|---|---|---|---|---|
|
Type |
Manufacturer |
Serial no.: |
|||
|
-Inv. no. |
Internal equipment no. |
||||
|
Site |
Operator |
||||
|
o New equipment since: |
o Equipment adopted since: |
o "Old equipment" |
|||
|
Action |
yes |
no |
not |
Date |
Comment |
|
Measuring points calibrated, adjusted, compared with display unit |
|||||
|
Return to "National Normal" carried out |
|||||
|
Function of the equipment checked based on the function description and ok |
|||||
|
Function of the alarms checked and ok |
|||||
|
Calibration and maintenance established (type, intervals, etc.) |
|||||
|
Recorded in maintenance schedule |
|||||
|
SOP "Operation and maintenance" compiled |
SOP no.: |
||||
|
Operating manual created/reviewed |
|||||
|
PQ required |
|||||
|
Comments/deviations: |
|||||
|
Evaluation |
|||||
|
The OQ was carried out by: |
The OQ was assessed and released by: |
||||
|
Name/Date ... |
Name/Date ... |
||||
Checklist for performance qualification of equipment/facilities
|
Equipment |
|||||
|---|---|---|---|---|---|
|
Type |
Manufacturer |
Serial no.: |
|||
|
-Inv. no. |
Internal equipment no. |
Equipment no.: |
|||
|
Site |
Operator |
||||
|
o New equipment since: |
o Equipment adopted since: |
o "Old equipment" |
|||
|
Action |
yes |
no |
not |
Date |
Comment |
|
IQ and OQ completed |
|||||
|
Equipment checked based on routine tests, which are described in operating procedures and test protocols and ok |
|||||
|
Function of the equipment checked within the limits and ok |
|||||
|
Equipment checked under near-real conditions by three consecutive production batches/analytical tests/measurements and ok |
1.: 2.: 3.: |
||||
|
Reaction of the equipment to improper/irrational operation remains plausible, the function of the equipment is not impaired |
|||||
|
Function is not impaired by entering values outside the limits/which exactly match the limit |
|||||
|
Comments/deviations: |
|||||
|
Evaluation |
|||||
|
The PQ was carried out by: |
The PQ was assessed and released by: |
||||
|
Name/Date ... |
Name/Date ... |
||||
1.C.6.4 Quality management system for validation
When companies speak of validation, in most cases they only mean process validation, i.e. the manufacturing process. However, the term validation also comprises the analytical methods, the cleaning of equipment and rooms and, possibly, the cleaning of the product itself. It is important to include the analytical techniques in the qualification/validation system, as, for example, it is a basic requirement for carrying out qualification that the measuring data be recorded with validated methods. During process validation, validated analytical methods are indispensable, as otherwise the information from the individual validation levels and the samples drawn can only be enjoyed with great caution.
When implementing a validation system, the Quality Unit must in principle establish two large systems in its procedure:
- Validation of the manufacturing processes and cleaning of equipment and rooms
- Validation of analytical methods
The basic structure of the quality management system is the same for both activities. They differ only in the way they are executed and in the target definition. However, as analytical methods are validated differently from the process validation, it is advisable in this case to compile two separate SOPs; one for the analytical methods, the other for the processes.
Prerequisites for validation
As with qualification, important basic requirements must be available before starting validation, without which no reasonable processing is possible. When compiling a validation protocol, these points are to be formulated as necessary requirements and should be inserted in a defined form for the protocol by the Quality Unit:
- Qualification of the equipment and facilities used is completed and documented.
- The analytical methods used are validated.
- The process is established in a valid master formula manufacturing/ instruction.
- The quality of the raw materials is established for the validation studies.
- The process-specific validation protocol is available and formally approved.
- The change control procedure for processes, facilities and methods is established and used.
- The responsible persons have been appointed.
For more information on qualification and validation, see chapter 6 Qualification and chapter 7 Process Validation.
|
Summary Qualification and validation make a significant contribution to the safety and reproducible quality of the products. However, it is important for the individual stages of the qualification and validation sequence to be interlocked in an expedient manner and processed in the necessary sequence. |
1.C.7 Training
|
Here you will find the answer to the following question: How is an expedient training system implemented? |
The qualification of staff is crucial for the quality of the manufactured products. This is because the procedures in the pharmaceutical industry are not, as is the case in many other industry branches, highly automated, but require various manual activities in nearly all manufacturing steps. This includes:
- a monitoring function, such as complying with temperature and time
- visual checks of individual steps (colour, appearance, etc.)
- manual activities such as weighing raw materials, mixing components, cleaning equipment or rooms or combining individual equipment components
In many procedures, other steps also have to be carried out, which require a special knowledge or skill for the respective activity, which is not learnt during professional training. So, in addition to the professional training of the staff employed in manufacturing and quality control, regular further training is also necessary. This also ensures adequate communication of the important and quality-critical points and processes in the event of internal changes or when new activities are commissioned.
It is therefore clear that the complex of training courses is split into two areas:
- General further training
- Training on specific problems and processes
General further training usually concentrates on the GMP requirements, and is to be carried out in regular intervals. The specific training courses have more complex contents and subjects and consist of points that relate to the sequence of daily work. They should be based on the problem, depending on necessity and focus.
1.C.7.1 Compilation of a standard operating procedure (SOP)
In order to group together and coordinate the not insignificant number of training measures for the company's staff in respect of new or changed regulatory requirements, the following points should be stipulated in an SOP by the Quality Unit:
- Compilation of a training program for the coming year:
- Demand/request for necessary training courses
- Compilation of the required training courses
- Requests in the departments (form)
- Approval by the management board
- Coordination of comprehensive training courses:
- Merging similar courses from different departments and areas
- Definition of dates
- Carrying out the training
- by the Quality Unit, internal or external experts
- Checking the efficacy of training courses:
- by the trainer/Quality Unit
- Documentation of the training carried out:
- Participant list
- Training documents
- Compilation of certificates
- Guaranteeing participation
1.C.7.2 Compilation of an annual program
For the compilation of a training program for the coming year, subjects are determined from the specialist departments. The Quality Unit and the further training department add the points which, from their perspective, require training. Using all these proposals, the Quality Unit, with the help of the further training department, compiles the final training program which takes into account all the requirements of the company as a whole.
This is forwarded to the company management for approval, as time and resources (budget) have to be provided just like for inspections. The further training plan should include subjects from the previous year which were not processed. As the Quality Unit receives end of year data on further training measures that were carried out or are still open for its report to the management board, it can also add the open measures. The Quality Unit distributes the further training plan approved by the management board to all departments and areas.
1.C.7.3 Compilation of a training plan
In collaboration with the further training department or the internal/external trainer, the Quality Unit should compile a training plan in good time, which gives detailed information on the structure of the respective training course. This is distributed to the participants of the course in good time, so that they are clear about the content and objectives.
Training plan points can include:
- Subject of the course
- Name of the trainer
- Location
- Time sequence:
- Start of the course
- Breaks
- End of the course
- Subjects of the individual training points
1.C.7.4 Guaranteeing participation
When establishing training courses and the scope of training, the group of people who must participate in the training course, must be specified. In this context, it must be ensured when holding and documenting the training course, that all staff who are obligated to participate in the training, were also actually present. In many cases, it is necessary to offer a repeat date, as, for various reasons, it is not always the case that all the designated people will have attended.
For more information on training, see chapter 2.C Training.
|
Summary Next to staff motivation, training is an essential element of personnel management, ensuring that the work to be carried out achieves the necessary and expected quality. Regular training courses ensure a specific level of certainty in daily work and in any problem situations that may occur. |
1.C.8 Inspection
|
Here you will find the answer to the following question: How is an efficient inspection system structured? |
According to chapter 9 of the EU GMP Guideline, all companies are obligated to carry out self-inspections. These are inspections that are carried out within the company by internal auditors. It is usually the employees of the Quality Unit who carry out the inspections.
In addition, there is the further requirement that the suppliers of raw materials, products or services be audited initially upon starting contractual relationships and thereafter in regular intervals. In this case, the company itself may be a supplier and must therefore be audited by an external auditor. At the same time, the company may make use of external help, in which case it is itself obligated to audit the suppliers or service providers based on a carefully executed risk analysis.
In principle, two systems must be established:
1. one for carrying out audits (internally and at external companies)
2. one for preparing and supporting audits by third parties in own company (authorities and contract givers)
1.C.8.1 Compilation of a standard operating procedure
For the SOP self-inspections and inspections to be carried out externally, the points listed in figure 1.C-17 must be defined.
|
Contents of an SOP inspection |
|---|
|
|
Extension for external inspections SOP:
|
1.C.8.2 Contents of the audit program
DIN EN ISO 9000:2000 gives the following definitions
- Audit program (chapter 3.9.2): Set of one or more audits (3.9.1), which are planned for a specific period of time and have a specific purpose.
- Audit plan (see chapter 1.C.8.3 Contents of an audit plan): Description of the activities and regulations on-site for an audit
At the start of a year, an audit program is compiled which contains all inspections planned for the period under review. This is compiled by the Quality Unit in collaboration with the individual specialist departments and is signed and released by the management board.
The following points are defined in the audit program:
- Time of audit
- Audited department/area/company
- Lead auditor/Audit team
- Audit objective/regulatory basis/focuses of the audit
- Time of audit
- Compilation of the audit report
- Audit completed
- Comments
This audit program can be used in several respects:
- The structure of the audit system can be shown to external auditors using this document and the above-mentioned SOP. For the authorities, this paper is used to prove that audits/self-inspections were planned, carried out and completed.
- Internally and externally, the document is used to inform the departments, areas or customers of the planned audit, so that they can prepare specifically.
- The members of the audit team are informed of their deployment in the planned audits in good time and can thus schedule these dates in advance.
- For the company management, it is an important indication of the costs and resources that will be involved in the inspections over the course of the year.
- The company management becomes involved by signing the audit program. In doing so, it accepts responsibility for the functionality of the quality management system.
- When compiling the new annual program for the following year, the audits that were not carried out can be carried over by the Quality Unit.
- The monitoring of dates for the regularly planned inspections/self-inspections can be carried out using this document.
- The columns: "Compilation of the audit report" and "Audit completed" can also be used by the Quality Unit to monitor dates for the current inspections.
1.C.8.3 Contents of an audit plan
The audit plan itself is the name for the flow chart for a specific audit, in which the times, focuses and people to be audited are listed. In order to prepare for the inspection as well as possible both on the part of the auditor and on the part of the auditee, the lead auditor sends the audit plan to the department, area or external company to be audited. The following points should be included in the audit plan (see figure 1.C-18):
|
Audit Plan Company XXX |
||
|---|---|---|
|
Audit date: Time: |
21. 06. 2004 - 22. 06. 2004 8:00 am - approximately 5:00 pm on both days |
|
|
Objective of the audit : Audit criteria: |
Approval as supplier Proof of fulfilment of requirements in accordance with SOP: supplier selection |
|
|
Reference documents: |
EU GMP Guideline |
|
|
Area(s) to be audited: |
Manufacturing, quality control, storage, raw material goods receipt |
|
|
Persons involved: |
||
|
Company XXX: |
|
|
|
Auditor: |
Head of Quality Unit |
|
|
Documents: |
SOPs, Manuals, manufacturing documents, control documents, proof of storage, validation documents |
|
|
Day 1 |
||
|
Time |
Subject |
Participant |
|
8:00 am - 8:30 am |
Welcome, introduction of the participants, establishment of the audit plan |
all |
|
8:30 am - 10:00 am |
Quality assurance system |
Head of Quality Unit/quality assurance |
|
10:00 am - 12:30 pm |
|
|
|
12:30 pm - 1:30 pm |
Lunchtime |
|
|
1:30 pm - 2:30 pm |
Dissolution of the raw material |
|
|
2:30 pm - 6:00 pm |
|
|
|
6:00 pm - end |
Concluding discussion Day 1 |
|
|
Day 2 |
||
|
8:00 am - 8:30 am |
Introduction day 2 |
|
|
8:30 am - 11:00 am |
Autoclaving of the product |
|
|
11:00 am - 12:30 pm |
Quality control and release |
|
|
12:30 pm - 1:30 pm |
Lunchtime |
|
|
1:30 pm - 2:30 pm |
Determination of the conditions/processes for possible purchasing |
|
|
2:30 pm - 4:30 pm |
Validation of the procedures |
|
|
4:30 pm - 5:00 pm |
Concluding discussion |
all |
For more information on inspections, see chapter 18 Inspections.
|
Summary In order to structure the inspections effectively, be it internally or at external companies, they should be planned and carried out carefully. This includes the compilation of an audit program, in which all planned audits and inspections for a year are listed. An audit plan describes the execution of an audit in detail (where, who, when, what). |
1.C.9 Batch record review and annual product review
|
Here you will find the answer to the following question: What quality management system is to be established? |
1.C.9.1 Batch record review
After all manufacturing steps have been carried out and after the analytical testing of all raw materials and manufacturing steps, the compiled documentation must be inspected before the release of a specifications-compliant batch. Simply filling in and signing all forms and documents is not sufficient to release the drug product after the final tests by quality control have been passed.
The EU GMP regulations specify that all batch records be assessed by quality control. These documents must be reviewed before the start of the analytical testing of the final product. Any deviations, changes or even faults, which have been documented but did not directly stand out, can be assessed by quality control and taken into account in the scope of the sampling, the sample volume and the assessment of the results of the final test. If quality control is integrated in the Quality Unit, then the batch record review can be carried out by the Quality Unit. The head of quality control is then notified of the result so that the corresponding work can be initiated, if necessary.
The batch record review is required in the bodies of rules and regulations, but in most cases the peripheral documents are not included. However, it is just as important to include these as to review the manufacturing process itself. When checking the peripheral documents, the Qualified Person must establish a system that ensures that he receives all the necessary information before certification of the batch. To this end, he can use the documents that are compiled for the hygiene of rooms, machines, facilities and personnel, the validity of the procedures and the stability of the products.
Another important point is the archiving and the associated storage time for documents. Manufacturing documents, together with the quality control documents, must be stored for at least five years (sometimes longer - e.g. documents from change management, deviation and OOS system, audit reports, data from trending and important records). In the event of any complaints or grievances by the customer, it must be possible to review the relevant manufacturing documents again. It would be fatal if an inconsistency or error was found in the documents at exactly this time. Faultless batch documentation, on the other hand, takes this burden off the company.
This means the manufacturing documentation review can also be used to find and eliminate any problem areas or faults that were not directly discovered during the product analysis.
An SOP must regulate who provides which documents at what time. As all areas must give this information to the Quality Unit, responsible persons are to be appointed who must send the entire documentation to the Quality Unit after completion of manufacturing.
In addition to the batch records, the extended documentation should also be considered, from which valuable additional information can be drawn in many cases. Specifically, the EU GMP Guideline does not contain any further information on the scope and depth of the Batch Record Review. Therefore, it is advisable to draw the corresponding requirements from the other bodies of rules and regulations. For example, support can be obtained from the Qualified Person's tasks in terms of the release (see Annex 16 of the EU GMP Guideline, chapter C.6.16 Annex 16 Final Version: Certification by a Qualified Person and Batch Release). In principle, in accordance with the regulatory requirements for the certification of products, the Qualified Person must review all documents which are directly or indirectly related to the production of the product and thus with the product quality, before sales.
It is the Quality Unit's task to use a system to structure the review effort for the company in an acceptable manner, to comply with the requirements made, not to forget any necessary measures or to implement unnecessary processes.
Support of the Qualified Person
The sales product for release must be certified by the Qualified Person. As in most cases they will not be in a position to view and assess the entire required documentation before certification, support can be provided by the staff of the Quality Unit. It is important that the Qualified Person is convinced that the staff can carry out the necessary work in a trustworthy fashion and that he can absorb the data. For this purpose, the Qualified Person should train the persons who will carry out the Batch Record Review in the Quality Unit. The course should stress the importance of the review and the exact consideration of the critical points in particular (possibly name them).
To make the Quality Unit's task easier, a checklist can be used for more extensive documentation, in which the critical points which must be reviewed under all circumstances are always checked in the review. To this end, a selection of further points can be added statistically, which then give a general overview of the complete and correct documentation. The Qualified Person must define requirements for the checklist based on a risk analysis together with the current changed requirements for the extended, statistical review of the documentation.
Carrying out the Batch Record Reviews
When inspecting the documentation, it is not sufficient to pay attention only to the completeness of the entries and signatures of the documents. The calculations made should also be repeated here and there. Plausibility checks can also detect problems in some cases. In this context, it is absolutely necessary to make use of the raw data.
In order not to let a routine develop for the employees carrying out the work, it is also advisable to regularly alternate the documents to be reviewed by the respective persons carrying out the review. When reviewing the documents it must be ensured that - particularly in the Quality Unit - the persons who compiled the documents are not responsible for reviewing them. The best solution is if the Qualified Person receives the documents for review from the Quality Unit, as they deal with the quality management system and give a good overview of the system.
For more information, see the GMP Manual chapter 15.C.5 Batch record review.
1.C.9.2 Annual product review
In order to be able to guarantee a consistently high product quality, it is necessary to critically assess the produced quality of the sales products in relation to manufacturing and quality control in regular intervals. This should be assessed at least once a year for each product.
In the same frequency, it is also checked if the objectives set for the quality management system have been fulfilled, such as
- Target agreements,
- Training program,
- Audit program,
- Trending,
- Monitoring,
- Management review.
The results from the previous year together with the outlook for the following year give a general overview of the status of the implemented quality management system. In America, this linking of the two sets of results is called the "Annual Product Review". In Europe, the EU GMP Guideline uses the term "Product Quality Review". The responsibilities and process of a "Product Quality Review" are to be established in an SOP. The Quality Unit, as the central position, should request this review and coordinate the necessary work. It defines which information is to be delivered in which format and scope from the individual departments and areas, so that a summary of all relevant data and results can be made in the Quality Unit.
Further information is provided in chapter 15.F Annual product review.
|
Summary The batch record review is an important measure before carrying out the analytical tests and in particular before releasing the product for sale. It supports the work and responsibility of the Qualified Person, in particular in this last point. In addition, it largely guarantees the correctness of the documents to be archived. |
1.C.10 Qualification of suppliers and service providers
|
Here you will find answers to the following questions:
|
In a pharmaceutical company, external suppliers and service providers play a crucial role in the manufacturing and quality control process. There is scarcely a company that produces all starting materials itself or does not use any external services. For example, the maintenance and servicing of complex facilities and machines is often carried out by external companies. The costs of holding the capacities in-house, in order to be fully autarkic, would be very high. Likewise, the company relies on suppliers and service providers during the manufacturing of preliminary and intermediate stages or of drug products. In order to ensure a consistently high quality of the services and starting materials supplied by suppliers, a system must be established to qualify them. The procedure and general understanding are to be codified in an SOP.
Figure 1.C-19 shows an example of an SOP for the selection and monitoring of suppliers and service providers, which can be extended or changed as required.
|
Example SOP: Selection and monitoring of service providers and suppliers |
||
|---|---|---|
|
Compiled by: |
Name (function) |
Date |
|
Checked: |
Name (function) |
Date |
|
Checked: |
Name (function) |
Date |
|
Approved: |
Name (function) |
Date |
|
Released: |
Name (function) |
Date |
|
Reason for compilation/change: |
Compiled again for purpose of defining internal standards |
|
|
Supporting documents: |
Purchasing guideline xxxxx |
|
|
1. Contents 2. Purpose of the document page 47 3. Scope page 47 4. Responsibilities page 47 5. Selection and monitoring of suppliers for materials that directly 5.1 Selection and approval page 48 5.2 Current monitoring of suppliers page 49 6. Selection and monitoring of service provision companies page 49 6.1 Selection and monitoring of EDP service providers page 46 6.2 Selection and monitoring of consultancy firms page 46 Appendix 1: Approval of an existing supplier Appendix 2: Approval of a new supplier Appendix 3: Supplier assessment for product-quality related materials |
||
|
2. Purpose of the document: This document gives detailed information on the procedures and responsibilities for the selection, approval and monitoring of suppliers and service providers. However, this does not relate to the selection and monitoring of contract manufacturers. The definition of the procedures should achieve the following objectives:
|
||
|
3. Validity These regulations apply for the entire field of company ... |
||
|
4. Responsibilities Every employee involved in the procurement of materials and/or services is responsible for complying with the requirements described herein. The responsibilities for subtasks are established as follows: |
||
|
Subtask |
Responsible person |
|
|
Obtaining suppliers' self-disclosure |
Respective purchasing officer (using quality assurance checklists) |
|
|
Audit |
Head of quality assurance |
|
|
Approval of supplier |
QA team |
|
|
Contract with the supplier |
Legal department |
|
|
Reporting of deficiencies to the quality assurance and complaint |
Purchasing representative |
|
|
Documentation on suppliers |
Purchasing representative |
|
|
5. Selection and monitoring of suppliers for materials that directly influence the product quality Such materials are, for example:
|
||
|
5.1. Selection and approval Suppliers for these materials are selected as follows:
|
||
|
||
|
5.2 Current monitoring of suppliers for materials that directly influence the product quality Suppliers for these materials are monitored as follows:
|
||
|
6. Selection and monitoring of service provision companies |
||
|
6.1 Selection and monitoring of EDP service providers The selection of suppliers for EDP services/software is undertaken by the EDP management in agreement with the respective purchaser/specialist department responsible for the application, in accordance with the following criteria:
In the current business relationship, the EDP management primarily monitors the reliability and efficiency in the event of complaints and problems. The list of qualified EDP suppliers is compiled and continuously updated by the EDP management. |
||
|
6.2 Selection and monitoring of consultancy firms Consultancy firms are selected as follows:
|
||
1.C.10.1 Responsibilities
The Quality Unit has overall responsibility for the qualification and monitoring of suppliers and service providers as well as for carrying out all established steps and the corresponding documentation. Responsibilities, together with specific activities for this process, must be clearly defined in order to avoid uncontrolled assessments, approvals and hasty activities without prior release. Final responsibility for the approval of suppliers and service providers should lie with the legally responsible person, as, in almost all cases, the quality of the starting materials and services is ultimately entered in the application file for marketing authorisation, and the validity of the procedure is affected.
For subtasks, the responsibilities can be defined as described in figure 1.C-16, point 4.
1.C.10.2 Risk analysis for grading
Suppliers and service providers cannot be treated in the same way, as the influence on the quality of the sales product differs depending on the type of the work to be provided or the starting material. This influence on the product quality is determined using a carefully managed risk analysis. Based on the results of this analysis, suppliers and service providers are to be qualified accordingly. In the starting phase of system implementation, it is recommended that you primarily assess the new service providers and suppliers. In the second phase, the risk analysis is extended to the established suppliers and service providers. Based on the risk classification, qualification can then be carried out together with monitoring of the suppliers and service providers.
|
Criteria for the risk analysis could be: |
|---|
|
Risk group I: Supplier/service provider has a direct influence on the product quality, such as:
|
|
Risk group II Supplier/service provider has an indirect influence on the product quality, such as:
|
|
Risk group III Supplier/service provider has no direct influence on the product quality:
|
1.C.10.3 Carrying out
The individual qualification steps (see figure 1.C-21) are described in detail in a form.
|
Qualification steps: |
|---|
|
The difference in carrying out qualification of companies that are graded in individual risk groups lies purely in the level of processing of the individual points. If suppliers and service providers are graded in risk group I based on the risk analysis, then all the above-mentioned points must be dealt with in detail.
In certain cases, however, inspection of the supplier or service provider in risk group I can be dispensed with, in order to save internal resources. This includes, for example:
- The supplier or service provider in question also works for other large and important pharmaceuticals companies.
- They are monitored by the authorities.
- They have recently demonstrably already survived several audits - possibly receipt of the latest or any audit report.
- There is collaboration at other levels (committees, other departments or areas in own company, etc.).
- They sell EU-certified materials (pharmacopoeia certificates).
In this context, it should be mentioned that no body of rules absolutely specifies that an audit be carried out. However, it is inferred that the supplier or service provider should grant the contract partner the possibility of carrying out an inspection. In a supplier or service provider audit, it is usually possible to solve many questions and problems.
It is important that the Quality Unit prepares the decision for the legally responsible persons to the extent that they can reach a satisfactory decision. Figure 1.C-22 gives an example of the approval of a supplier.
|
Approval of a new supplier |
||
|---|---|---|
|
Supplier: ... |
||
|
For product/product group: ... |
||
|
Assessment of the available information: ... |
||
|
Written promise by the supplier, that the product meets the specifications of "GMP-Pharma" in all points |
Available |
|
|
Missing |
||
|
Safety data sheet |
Available |
|
|
Missing |
||
|
Not applicable |
||
|
Sample checked by: ... |
Ok |
|
|
Defective |
||
|
Not applicable |
||
|
Written declaration of consent that "GMP-Pharma" can carry out an audit at any time |
Available |
|
|
Missing |
||
|
Written promise that no changes will be made to the product to be supplied without prior written consent of the "GMP-Pharma" |
Available |
|
|
Missing |
||
|
Supplier self-disclosure |
Available |
|
|
Missing |
||
|
Sufficient |
||
|
Inadequate |
||
|
Guarantee of delivery in the specified time frame (particularly important for repacking actions) |
Available |
|
|
Missing |
||
|
Audit |
Positive |
|
|
Deficiency |
||
|
Negative |
||
|
Not necessary |
||
|
Based on this information, the supplier is graded as: |
Approved |
|
|
Conditionally approved |
||
|
not permitted |
||
|
The assessment was carried out in the QA team on ... ______________________________ (Head of QA team) |
||
1.C.10.4 Requalification
After initial approval as a qualified supplier or service provider, requalification should be carried out in regular intervals, based on data from the fields of
- Purchasing,
- Production,
- Quality control,
- Quality Unit,
- and possibly other areas (depending on the product/service).
The maintenance of the qualification should be described and listed in the "Qualification of Suppliers and Service Providers" SOP. Data that can be used for requalification are shown in figure 1.C-23, for example.
In the pharmaceutical area, the intensive monitoring of suppliers and service providers can be restricted to companies who are in risk group I. However, the suppliers and service providers in risk groups II and III must also be monitored an requalified. The procedure for these risk groups, however, is to be adapted accordingly.
The trend of suppliers and service providers in risk class II gives information on whether or not the quality of the materials or work deteriorates in the course of time. Accordingly, counteractive measures must be taken to ensure that the certified status is achieved again. The final consequence is to change the supplier or service provider.
In the annual review of the quality management system for the management review, data from the supplier assessment can also be recorded and processed.
In many cases, external suppliers and service providers make a not insignificant contribution to helping a pharmaceutical company provide high quality products. The legally responsible persons should be aware that their pharmaceutical responsibility for the product is associated with the activities of the many suppliers and service providers in nearly all cases. The intended completion of a pharmaceutical responsibility delimitation agreement by the quality management system is absolutely necessary
|
Requalification of suppliers for quality-related materials (risk group I) |
||
|---|---|---|
|
Supplier: ... |
Date of assessment: ... |
|
|
Product/product group: ... |
||
|
Number of deliveries since last assessment: .... |
||
|
Quality parameters: |
||
|
Supplied material compliant with specifications: |
Always |
|
|
Occasional problems/complaints |
||
|
Frequent problems/complaints |
||
|
QA system installed |
Yes, in accordance with: ... |
|
|
No |
||
|
Yes, not certified |
||
|
Yes, certified in accordance with DIN EN/ISO ... |
||
|
Audit carried out |
No, reason: ... |
|
|
Yes, result: |
||
|
||
|
||
|
||
|
Behaviour regarding quality complaints: |
Very cooperative |
|
|
Averagely cooperative |
||
|
Uncooperative |
||
|
No complaints in the period under review |
||
|
Reliability: |
||
|
Quantity received is same as order quantity |
Always |
|
|
Occasional deviations |
||
|
Frequent deviations |
||
|
Delivery date ±3 days complied with |
Always |
|
|
Occasional delays |
||
|
Frequent delays |
||
|
Condition of delivery ok (containers, cleanliness, labelling) |
Always |
|
|
Occasional problems |
||
|
Frequent problems |
||
|
Documentation ok (delivery papers, certificates, etc.) |
Always |
|
|
Occasional problems |
||
|
Frequent problems |
||
|
Willingness to cooperate: |
||
|
Behaviour during audit |
Open |
|
|
Passive |
||
|
Uncooperative |
||
|
Not relevant as no audit carried out |
||
|
Behaviour during commercial complaints |
Open |
|
|
Passive |
||
|
Uncooperative |
||
|
Availability of the contact person |
Good |
|
|
Sometimes problematic |
||
|
Inadequate |
||
|
Duration of the business relationship: ... |
||
|
Comments: ... |
||
|
After assessment of the above-mentioned data, the QA team suggests that the supplier ... for product/product group ... |
||
|
should continue to be graded in |
the approved class |
|
|
the conditionally approved class |
||
|
the rejected class |
||
|
Approval as supplier granted: |
no |
|
|
yes |
||
|
|
|
|
.
For more information, see chapter 18.F Inspection of contract manufacturers and chapter 18.G Inspection of suppliers.
|
Summary: Contract manufacturers and service providers make a significant contribution to the product quality, depending on the task. It is thus very important to carefully select good and reliable contract manufacturers and service providers. Regular verification of their reliability is just as necessary as a continuous review of the quality of their work. |
