|
Here you will find answers to the following questions:
|
"If something is not documented, it has not been carried out." This guideline shapes the entire documentation system of the GMP-regulated area of the pharmaceutical industry. Proving that a manufactured product is of the specified quality is an essential issue in the daily business of the pharmaceutical industry. Documents are always required which show that before manufacture, quality control and distribution of the medicinal product commence, the individual steps to be carried out were planned correctly and that all activities resulting from this have been carried out according to the specification.
The proof that the two requirements - planning and implementation - have been met can only be proved with adequate documentation.
The aim of documenting the manufacturing, control, storage and dispatch must therefore be for a company to be able to consistently prove at any time that a product sold is of the required quality using the recorded data.
1.B.1 Structure of a documentation system
Documentation is the central element in a pharmaceutical quality management system. It is therefore important to give this most important element a structure which in turn also reflects the structure and organisation of the implemented QM system.
Unfortunately, in discussions about documentation or a corresponding structure, we often recognise that it almost exclusively concentrates on proving that the medicinal product is manufactured and controlled in accordance with the specifications. During an inspection, it is the evidence of manufacturing and controlling of a medicinal product that is almost exclusively asked for. However, the structure in a company does not only include the documents that are created during active manufacture and for release at the end of production. After all, quality assurance represents an entire building and also includes areas and departments that are not directly involved in the manufacturing and quality control. It is therefore important that the system behind these final documents is also taken into consideration and its robustness, completeness and also its effectiveness checked and scrutinised. This is the only way to get an overview of the entire structure and to facilitate your understanding of many processes. In addition, it will allow you to recognise the extent to which the neighbouring areas of production and quality control are integrated in the GMP system.
The basic structure of the entire building can essentially be divided up as follows (see figure 1.B-1). On the left are the basic structures and on the right are the levels responsible for implementing the corresponding documentation.
|
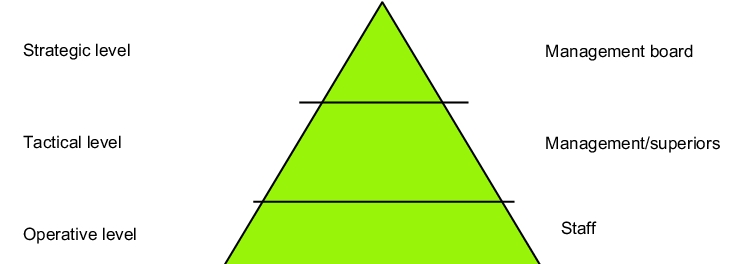
If we go through the individual levels systematically, we can recognise that the volume and the degree of detail increases significantly - as regards GMP relevance - from the top management board level down to the on-site staff.
EU GMP Guideline, Chapter 4, Documentation |
|---|
|
The question now arises as to which documents must be compiled and implemented in the individual levels listed. It is therefore necessary to name the individual documents so that it is understood consistently throughout the company. In accordance with the EU GMP Guideline, the following definitions, with regard to the manufacturing and quality control areas in particular, are used and should be adopted as they stand (see figure 1.B-2).
In the quality management system, the Quality Unit must determine which documents are compiled and also implemented at which hierarchical levels. This is essentially based on the basic structure listed above and should look as described below.
1.B.1.1 Management board level
It is necessary for the management board level to let the Quality Unit work out a system which specifies the documents that they are to compile and take responsibility for.
These include, for example:
- Quality management handbook/PIC site master file
- Management review
- Minutes of meetings
- Job descriptions
- Quality objectives
These are based on the considerations that are detailed in chapter 1.C.1 Management responsibility and the activities and responsibilities stated there. As you can see, the list is made up of more or less strategic documents that rarely require modification.
Structure specifications must be determined for the quality management handbook, the PIC site master file, the job descriptions and quality objectives.
Most importantly, these include specifications such as:
- the composition,
- the external appearance,
- the content,
- the approval process,
- distribution and
- the revision procedure
The necessary specifications can be set down as a template in a corresponding SOP or can be included in the quality management handbook as an appendix or descriptive specification. Not all the documents listed must necessarily be archived by the management board, but it must adopt the responsibility for their existence, completeness and currency. However, it goes without saying that the management board is at least familiar with the content of these documents. Normally, the documents are in the care of the Quality Unit, where they are kept up to date and any change is discussed with the management.
In particular for generally applicable documents, it should be demonstrated that the staff in the company are familiar with the content and are aware of the requirement to specify and describe their day-to-day work. The Quality Unit must develop a system that guarantees the currency and validity of the documents on the one hand and which ensure their communication and the staff's comprehension of the content on the other hand.
1.B.1.2 Management/superiors
At the next level, there is the responsibility for compiling and approving all the documents that contain a description or a general overview of the day-to-day work. They are mostly descriptive or have strategic significance. They include the following documents, for example.
General operating procedures, such as:
- Master formulae - in the aforementioned sense - for manufacturing and control
- Maintenance and calibration of apparatus
- Qualification/validation of apparatus and procedures
- Change Management
- Deviation management
- Plans and reports of activities that support the aforementioned instructions
- Specifications
- Maintenance and monitoring of facilities, apparatus, equipment and rooms
- Flow charts for manufacturing and/or quality control
- Description of IT systems
- Procedures for dealing with service providers
Generally speaking, all documents that describe the work in general terms and/or provide generally-applicable instructions that the staff does not directly require for its day-to-day work should be located in the management level and come under its responsibility.
1.B.1.3 Staff level
This includes documents that describe in detail and specify the implementation of day-to-day operations, such as manufacturing, control, cleaning, maintenance, clothing changes, environmental monitoring, sampling, testing, use of apparatus, etc. The records resulting from this document the work carried out in accordance with the specifications. Documents for the staff can be divided into two groups based on their content:
- Specification documents: These are documents which specify the activities to be carried out and in which the work/result of the work must be entered, if required. The input fields are empty (instructions or SOPs - Standard Operating Procedures - in the original sense of the term).
- Record documents These are documents in which the activities carried out must be documented and proven with a signature and date (records).
1.B.1.4 Quality unit documents
The Quality Unit acts tactically when documents are compiled and operatively for other papers.
It has tactical duties to fulfil for quality management specifications. These may be in the form of specifications for structures, appearance and content of documents. For example, the implementation of the requirements is documented by definition in the "master SOP" and the SOP for the "numerical code". For a further example, see the specification for compiling scientific reports from the Research and Development area. These reports are required for compiling dossiers for the registration of medicinal products, for instance.
As well as the specification for the processes in the individual areas, the Quality Unit must also compile specifications for the tasks in its own area and document the activities carried out. For example, the carrying out of inspections, the processing of deviations or the implementation of the CAPA measures can be detailed here.
The specification documents that must be compiled by the Quality Unit include the elements of the quality management system itself:
- Master instruction/SOP
- Numerical code
- Administration/archiving
- Audits
- Training
- Qualification of equipment and facilities
- Validation of procedures/methods
- Complaints/returns processing
- Change management
- Selection/approval of suppliers
- Implementation of clinical trials
- Manufacture of drugs for clinical trials
- Batch record review/release
- Corrective and Preventive Actions (CAPA)
- Trending
- Selection/approval of service providers
- Checking the efficacy of training
- Risk analysis
- Specifications for the registration documentation
The list cannot be exhaustive and must be supplemented or modified as necessary, depending on the company and areas of expertise. It is only intended to show that a considerable number of SOPs must also be compiled by the Quality Unit.
1.B.1.5 Procedure description and procedure instruction
As well as the definitions from chapter 4 of the EU GMP Guideline, many companies also use two further designations that relate to the tactical level (procedure description) and the operative level (procedure instruction). The difference between the two designations can be seen in the words themselves. The procedure description describes a procedure to be carried out and the processes that it involves. The procedure instruction shows the staff how to carry out certain activities. In many cases, this conventional document is also called an SOP, as it is actually meant in American English (Standard Operating Procedure).
These two types of document have different target groups. The procedure descriptions are intended to provide appropriate information about the processes to external people who wish to be informed about company procedures.
These are, for example:
- External collaboration partners
- Authorities
- Auditors
- Regulatory authorities
- Other internal areas or parts of the company
Classic examples of this are the description of manufacturing and the implementation of the analytical tests in the control laboratory.
The target group for the procedure instructions is the on-site staff that actually implements the individual steps of the specified processes and then documents them in the attached records or in other defined places.
Generally, the structure of both documents is initially identical and follows the requirements of the master SOP. They only differ in the actual content and statements. It should be emphasised that the procedure descriptions are of an explanatory nature and also usually contain procedural diagrams (Flow Charts) to aid understanding.
However, there are also documents which represent a mixture of the two requirements. These documents are more frequently found in the Quality Unit and comprise nearly all systematic instructions, as for example:
- Change Management
- Deviations
- OOS
- Carrying out the inspections
- Complaint processing
- etc.
Examples are listed in the appropriate chapters ( to ) which show a flow chart together with a record as well as a description of the corresponding processes.
1.B.1.6 Operating procedure
Operating procedures play the decisive role in the day-to-day work of a pharmaceutical company (see also chapter 15.D Standard operating procedures (SOPs)).
A question which is often asked in relation to a quality management system and its documentation is how many SOPs are required. This question can never be satisfactorily answered. Each company must determine the number of SOPs to be compiled itself. However, the SOPs must be used to ensure that all the processes and associated activities are determined in specification documents, carried out accordingly and entered in the record documents.
In addition, a company operating worldwide must also take into consideration the fact that the notion of the content and number of required SOPs varies greatly between the European authorities and the American authority (FDA). The FDA expects that all activities are recorded in an SOP and that its content and explanations are such that nearly anyone could carry out this work.
In Europe, there is another comprehension to the SOPs because there are known to be many very well-trained employees in a company who have either qualified by means of special examinations or by many years' work in the appropriate field. For this reason, many of the activities are not described in depth, as is expected by the FDA. However, to summarise, the SOPs must be drawn up so that the activities to be carried out are described in sufficient detail - depending on the level of knowledge of the personnel. All work must be able to be found in corresponding SOPs.
Training should only take place for SOPs that cover activities that are beyond the normal level of knowledge of the relevant members of staff.Finally, it should also be mentioned that not all processes must be determined in the operating procedures. If an adequate description can be provided in procedure instructions or in the quality management handbook, this is sufficient.
1.B.2 Documents required in accordance with GMP
The EU GMP Guideline stipulate specific requirements for all types of documents which must be incorporated in the company-internal quality management system and interpreted accordingly by specifications from the Quality Unit. Relevant requirements can be found in chapter 4 of the EU GMP Guideline (see figure 1.B-3).
Chapter 4, Documentation |
|---|
General requirements
|
The requirements for documentation given in the EU GMP Guideline therefore determine the scope of the quality management system to be implemented. The Quality Unit now has the task of implementing these requirements on the documentation system and of introducing uniform regulations for all departments and areas.
There should also be specifications for areas that are not directly part of the GMP-regulated areas, for example:
- Clinical research
- Research and Development
- IT area
- Marketing and sales
In many cases, they perform quality-relevant activities and it is therefore obligatory to compile and maintain corresponding report documents, particularly in the record area.
The following points regarding the organisation of a documentation system must be observed:
- Structure specifications for the documents to be compiled
- Procedure for the compilation itself and a review
- Authorisation of the compiled documents
- Distribution to the individual areas or to the corresponding persons
- Handling of the documents on site
- Change procedures for valid documents
- Archiving of all finalised and completed documents
- Any distribution to external offices (registration, inspection by authorities, contract manufacturers, etc.)
It is equally important to monitor day-to-day compliance. This may be in the form of self-inspection at regular intervals (see chapter 1.C.8 Inspection). The requirements for documentation should be determined by the Quality Unit in the form of instructions (SOPs) - partly also signed by the management board - which describe all the necessary points and specify the responsibilities in detail.
1.B.3 Quality management handbook
1.B.3.1 Site master file
It is important to obtain sufficient information in the run up to the inspection of a company so that the auditor can get an overall picture. To this end, the Pharmaceutical Inspection Convention drew up specifications for the standardised description of the activities of a pharmaceutical company in 1993 in the form of a Guideline. This overview is known as the site master file (SMF). The specifications for the content of the SMF are based on the main chapters of the EU GMP Guideline and require a description with some evidence of the activities carried out there.
This guideline (PI 008-1, from November 2002) has the primary objective of providing supervisory officials from the PIC/S countries with the necessary information to speed up the marketing authorisation for medicinal products in the relevant countries.
This SMF will provide them with concrete information about the implementation and status of the quality management system in the company with regard to the manufacture and quality control of pharmaceutical products. figure 1.B-4 shows an example of the contents for an SMF.
Contents for an SMF in accordance with PI-008 |
|---|
1. Information about the company 1.1 Brief information about the company 1.2 Pharmaceutical manufacturing activities 1.3 Other manufacturing activities 1.4 Address of the plant 1.5 Manufactured products 1.6 Description of the plant 1.8 Use of other support 1.9 Presentation of the quality management system |
2. Personnel 2.1 Organisation charts 2.2 CVs of legally responsible persons 2.3 Education of the members of staff/training 2.4 Health requirements 2.5 Hygiene requirements |
3. Premises and equipment 3.1 Premises 3.2 HVAC systems 3.3 Special areas 3.4 Water supply 3.5 Maintenance of rooms and supply systems 3.6 Equipment 3.7 Maintenance of production facilities 3.8 Qualification, validation, calibration 3.9 Plant hygiene |
4. Documentation 4.1 Compilation, updating and distribution 4.2 Other product quality documentation |
5. Production 5.1 Brief description of the production processes 5.2 Handling of starting materials, packaging material, intermediate products and finished products. 5.3 Rejected materials and products 5.4 Description of the process validation |
6. Quality control 6.1 Activities/responsibilities |
7. Contract manufacture and analysis 7.1 Determining contract manufacture and analysis |
8. Sales 8.1 Products sales 8.2 Sales documentation |
9. Self-inspection Description of the self-inspection system |
10. Appendices 10.1 Site plan of the company 10.2 Plan of manufacturing rooms and quality control 10.3 Master hygiene plan 10.4 Flow charts (of complex processes such as: manufacturing, change management, complaints, etc.) 10.5 List of the most important/systemic SOPs |
Recently, the demand for the SMF has increased further. The SMF is not only used as an initial source of information by the supervisory officers, but it is also increasingly being requested by other external companies before they enter into business relationships in the area of medicinal products.
Also in external companies that have a close collaboration with the medicinal product manufacturer (see chapter 1.C.10 Qualification of suppliers and service providers), the question is increasingly being raised of the extent to which an individual company description is useful for these companies as an SMF. In many cases, the suppliers and service providers are incorporated in the SMF of the company concerned. However, this has no great relevance for the external company in relation to other contractual partners.
Further information is provided in chapter 15.E Site master file.
1.B.3.2 Handbook in accordance with EN ISO 9001:2000
Many suppliers and service providers have also undertaken EN ISO 9001:2000 certification to demonstrate their customer quality management system and to generally prove the existence of a functional quality management system. As part of this certification process, the standard requires that the company concerned has a quality management handbook (see figure 1.B-5).
4.2.2 Quality management handbook |
|---|
The organisation must compile and maintain a quality management handbook containing the following: a) The field of application of the quality management handbook, including details and reasons for any exclusions, b) The documented procedures created for the quality management system or references to it and c) A description of the interaction of the quality management system processes |
The handbook based on the ISO standard has the objective of describing the quality management systems in a certified company. However, it cannot be directly compared with an SMF, as the SMF focuses on the product quality and its safeguarding. The ISO handbook has the quality management system itself as its objective, without product quality playing a significant role.
Many companies are therefore in the complex situation of having and maintaining an ISO handbook for certification and being listed in the SMF for their pharmaceutical customers or having their own SMF. This therefore means that they have to maintain two handbooks. Figure 1.B-6 shows an example of a contents in accordance with DIN ISO.
Contents of a QM handbook in accordance with DIN-ISO |
|---|
1. Contents |
2. Foreword 2.1 Basics of the quality management system 2.2 Objective of the quality management system 2.3 Company profile |
3. Basis quality management system 3.1 Structure of the company 3.2 Structure of the system 3.2.1 General 3.2.2 Regulatory basics 3.3 Documentation for the quality management system/document structure 3.4 Document hierarchy 3.4.1 General 3.4.1.1 Document controls 3.4.2 Quality management handbook 3.4.3 Instructions 3.5 Quality planning 3.6 Quality management processes 3.6.1 Primary and secondary processes 3.6.2 Interaction of processes 3.7 Archiving 3.8 Management responsibilities and activities 3.8.1 Company management responsibility 3.8.1.1 Quality policy 3.8.1.2 Management of resources 3.8.1.3 Management review 3.8.1.4 Evaluation and improvement of processes 3.8.1.5 Internal communication 3.8.1.6 Training 3.8.1.7 Inspections 3.8.2 Change Management |
4 Production of the quality management system 4.1 Procurement 4.1.1 Orders 4.1.2 Control 4.2 Manufacture of products 4.3 Quality control |
4.4 Management of records 4.4.1 Batch reports 4.4.2 Laboratory reports 4.5 Storage 4.6 Identification and traceability of product/product status 4.7 Customer property 4.8 Reprocessing/rework 4.9 Management of defective products |
5. Accompanying quality management system 5.1 Design/project management 5.1.1 Development plan 5.1.2 Development entries 5.1.3 Development results/evaluation 5.1.4 Development verification/validation 5.2 Qualification and validation 5.2.1 Manufacturing process and equipment 5.2.2 Computer systems 5.3 Maintenance 5.4 Environment 5.5 Complaints 5.6 Data analysis/trending 5.7 Customer management 5.7.1 Determination of requirements/communication with the customer 5.7.2 Evaluation of customer satisfaction 5.8 Product documentation 5.8.1 Documents for registration |
1.B.3.3 Combined handbooks in accordance with GMP and ISO
In the meantime, nearly all supervisory officers throughout Europe have come to believe that an ISO handbook with an enhanced description of the product quality safeguarding is a very useful source of basic information for an inspection. A breakdown in the part of the description in chapter 1 of the EU GMP Guideline based on the ISO requirements, followed by the missing specifications relating to the manufacturing and quality control as an extra chapter to the basic SMF, provides a very good overview of the company. In order to better present the implementation of the specifications from the SMF, a comparison matrix can be entered at the end of the handbook, for example (see figure 1.B-7).
All noted certification companies conform with this procedure, as long as the requirements of the ISO standard are set down in the handbook.
EN ISO 9001:2000 |
PIC/S Site Master File |
|---|---|
1. Contents |
|
2. Foreword |
|
2.1 Basics of the quality management system |
|
2.2 Objective of the quality management system |
|
2.3 Company profile |
|
3. Basic quality management system |
|
3.1 Structure of the company |
1. Information about the company |
3.2 Structure of the system |
|
3.2.1 General |
|
3.2.2 Regulatory basics |
|
3.3 Documentation for the quality management system/document structure |
4. Documentation |
3.4 Document hierarchy |
|
3.4.1 General |
|
3.4.1.1 Document control |
|
3.4.2 Quality management handbook |
|
3.4.4 Instructions |
|
3.5 Quality planning |
|
3.6 Quality management processes |
|
3.6.1 Primary and secondary processes |
|
3.6.2 Interaction of processes |
|
3.7 Archiving |
|
3.8 Management responsibilities and activities |
|
3.8.1 Company management responsibility |
|
3.8.1.1 Quality policy |
|
3.8.1.2 Management of resources |
2. Personnel |
3.8.1.3 Management review |
|
3.8.1.4 Evaluation and improvement of processes |
|
3.8.1.5 Internal communication |
|
3.8.1.6 Training |
|
3.8.1.7 Inspections |
9. Self-inspection |
3.8.2 Change Management |
|
4. Production quality management system |
|
4.1 Procurement |
|
4.1.1 Orders |
|
4.1.2 Control |
|
4.2 Manufacture of products |
5. Production |
4.3 Quality control |
6. Quality control |
4.4 Management of records |
|
4.4.1 Batch reports |
|
4.4.2 Laboratory reports |
|
4.5 Storage |
|
4.6 Identification and traceability of product/product status |
|
4.7 Customer property |
|
4.8 Reprocessing/rework |
|
4.9 Management of defective products |
|
5. Accompanying quality management system |
|
5.1 Design/project management |
|
5.1.1 Development plan |
|
5.1.2 Development entries |
|
5.1.3 Development results/evaluation |
|
5.1.4 Development verification/validation |
|
5.2 Qualification and validation |
|
5.2.1 Manufacturing process and equipment |
3. Premises and equipment |
5.2.2 Computer systems |
|
5.3 Maintenance |
|
5.4 Environment |
|
5.5 Complaints |
|
5.6 Data analysis/trending |
|
5.7 Customer management |
8. Sales |
5.7.1 Determination of requirements/communication with the customer |
|
5.7.2 Evaluation of customer satisfaction |
|
5.8 Product documentation |
|
5.8.1 Technical documentation |
|
6. (Other chapters, free choice) |
7. Contract manufacture and analysis |
7. Appendices |
10. Appendices |
1.B.3.4 Functions of the quality management handbook
The quality management handbook should be regarded as evidence of the implemented and functioning QM system for external companies and authorities. For this reason, the responsibility for the validity of the content and implementation in accordance with the regulations belongs to the management board. Together with the Quality Unit, they must ensure that all the described systems and requirements for safeguarding and guaranteeing product quality are implemented as described and defined. The handbook cannot and must not be treated as simply a formality.On the other hand, it is also used to communicate the requirements and the implementation specifications for the quality management system from the management board. In this way, all members of staff are presented with the structure and content of the QM system in writing.
For companies that have a global presence, it is used:
- as a guideline for all sites,
- to harmonise important communication lines and
- to synchronise the quality management systems at the individual sites
The quality management handbook must therefore be authorised by the management board. They must make sure that the content is known to, understood and implemented in all parts of the company. The content must be checked for its currency at regular intervals and - if necessary - updated. The Quality Unit can provide support in both these matters. The currency is continuously checked at both internal and external audits and must then be adjusted if necessary.
The implementation is interpreted when other systemic SOPs are determined or during meetings and training.
Summary: A documentation system is composed of the management board, the management and the staff levels. We also distinguish between procedure descriptions, procedure instructions and operating procedures, depending on the character of the specifications and their target group. The EU GMP Guideline lists the minimum documents that must be available. The Quality Unit itself specifies the structure of documents (master SOP) and describes the elements of the QM system. The quality management handbook describes the QM system. It can be organised according to GMP regulations (e.g. PIC Site Master File), according to DIN ISO 9001:2000 or a combination of the two. |
