Национальный технический университет украины «Кпи»
факультет биотехнологии и биотехники
кафедра промышленной биотехнологии
Реферат
По “Генетической инженерии”
Тема:
ПОЛИМЕРАЗНАЯ ЦЕПНАЯ РЕАКЦИЯ И МУЛЕКУЛЯРНО-ГЕНЕТИЧЕСКИЙ АНАЛИЗ
| Роботу проверил: Клетчак И.Р. |
Работу выполнил: студент V курсу ФБТ Группы БТ-81 Островной Д.В. |
Киев 2002
Оглавление
| стр. |
|
| 3 |
|
| 1.1. Перспективы практического использования ПЦР-диагностики |
4 |
| 1.2. Полимеразная цепная реакция |
5 |
| 1.3. Амплификация РНК |
8 |
| 2.Общие положения PCR |
9 |
| 2.1. Подбор праймеров |
9 |
| 11 |
|
| 2.1.2. Полиморфные последовательности-мишени |
11 |
| 2.2. Приборное обеспечение |
13 |
| 2.3. Число циклов |
14 |
| 2.4. Точность |
14 |
| 2.5. Модифицированные методы |
14 |
| 2.5.1. Постановка ПЦР с использованием “горячего старта” |
14 |
| 15 |
|
| 2.5.3. Полуколичественный анализ. |
16 |
| 3. Работа с образцами тканей |
18 |
| 3.1. Подготовка пробы биологического материала. |
18 |
| 3.1.1. Нативные ткани |
20 |
| 3.1.2. Фиксированные ткани |
21 |
| 3.1.2.1. Фиксирующие вещества |
21 |
| 3.1.2.2. Длительность фиксации |
22 |
| 3.1.2.3. Изготовление срезов |
22 |
| 3.1.2.4. Выделение ДНК из срезов |
23 |
| 3.2. Проведение ПЦР |
25 |
| 3.3. Спинномозговая жидкость (СМЖ) |
31 |
| 3.4. Ткани, замороженные в среде, обеспечивающей оптимальные условия резания при данной температуре (ОСТ) |
31 |
| 3.5. Архивные препараты |
32 |
| 4. Анализ ПЦР-амплифицированной ДНК |
33 |
| 4.1. Подготовка ПЦР-продуктов |
33 |
| 4.2. Гель-электрофорез |
33 |
| 4.3. Гибридизация ПЦР-амплифицированной ДНК по Саузерну |
|
| 4.4. Дот-блот-гибридизация ПЦР-амплифицированной ДНК |
36 |
| 5. Интерпретация результатов |
37 |
| 5.1. Выбор контролей |
37 |
| 5.2. Чувствительность |
38 |
| 5.3. Количественный ПЦР-анализ |
39 |
| 6. Загрязнения |
41 |
| 7. Применение ПЦР в клинической лаборатории |
42 |
| 8.Организация технологического процесса постановки ПЦР |
45 |
| 9.Устройство ПЦР-лаборатории |
47 |
| 10. Критерии выбора ПЦР-набора |
49 |
| 52 |
|
| 12. Denaturating Gradient Gel Electrophoresis |
54 |
| 13. Литература |
56 |
Впервые состав ингредиентов, входящих в реакционную смесь для постановки полимеразной цепной реакции, и основные принципы использования праймеров (коротких искусственно синтезированных молекул ДНК) для получения копий ДНК были описаны Kleppe с соавт. в 1971 году. Однако тогда еще не была продемонстрирована основная черта ПЦР - экспоненциальное увеличение количества копий фрагмента исходной ДНК как результат реакции.[10] Это было осуществлено в 1985 г. на фирме Cetus. Последующее использование в ПЦР термостабильной ДНК-полимеразы существенно расширило возможности ее применения, как в научных целях, так и в клинике.[1] В 1985 году Saiki с соавт. опубликовали статью, в которой была описана амплификация геномной последовательности b -глобина. С этого момента количество публикаций, в которых авторы сообщали о применении ПЦР в своих работах, стало увеличиваться в геометрической прогрессии. Метод стал настолько популярен, что сегодня уже трудно представить работу в области молекулярной биологии без его использования. Особенно бурное развитие метод полимеразной цепной реакции получил благодаря международной программе «Геном человека». Были созданы современные лазерные технологии сиквенирования (расшифровки нуклеотидных последовательностей ДНК). Если в недавнем прошлом для расшифровки последовательности ДНК размером в 250 пар нуклеотидов (п.н.) требовалась неделя, то современные лазерные сиквенаторы позволяют определять до 5000 п.н. в день. Это в свою очередь способствует значительному росту информационных баз данных, содержащих последовательности ДНК. В настоящее время предложены всевозможные модификации ПЦР, показана возможность создания тест-систем для обнаружения микроорганизмов, выявления точечных мутаций, описаны десятки различных применений метода.[10] Мы вкратце рассмотрим теоретические основы ПЦР и применение этого метода для молекулярно-генетического исследования образцов тканей. Основное внимание будет уделено практическим аспектам, важным для анализа обычных тканевых образцов.
1.1. Перспективы практического использования ПЦР-диагностики
По данным журнала LabMedica International с 1995 года наблюдается увеличение расходов на ПЦР-диагностику, что является показателем роста использования ПЦР в практической медицине. Американские ученые отмечают, что в 1996 г. на ДНК-диагностику было потрачено 181.6 миллионов долларов, а к 2003 году, по прогнозам экспертов, затраты возрастут до 1400 миллионов долларов.
По данным того же журнала в настоящее время наиболее быстро развиваются пять основных направлений генодиагностики:
диагностика инфекционных заболеваний;
диагностика онкологических заболеваний;
- диагностика лейкемий и лимфом;
- диагностика рака груди;
- диагностика других злокачественных заболеваний;
диагностика генетических заболеваний;
идентификация личности;
- судебная медицина, криминалистика;
- трансплантация органов и тканей;
- определение отцовства;
диагностика патогенов в пище.
В отличие от иммуноферментного анализа, который широко используется для диагностики инфекционных заболеваний, ДНК-диагностика позволяет определять непосредственно возбудителя заболевания. С помощью усовершенствованных схем постановки ПЦР можно выявлять патогенные микроорганизмы в очень низкой концентрации.
ДНК-диагностика рака ограничивается небольшим, но активно возрастающим количеством сведений о генах, ассоциированных с раком. В рамках проекта «Геном человека» ученые продолжают поиски мутаций, ассоциированных с этим типом заболеваний.
Диагностика генетических заболеваний, также как и раковых, может развиваться только вслед за проведением широких научных исследований генома человека. Однако медицинское сообщество уже осознало важность изучения генетической основы заболеваний, а также возможность диагностирования и начала лечения болезни до появления ее симптомов.
Эксперты оценивают это направление на рынке ДНК-диагностикумов как одно из наиболее крупных и быстрорастущих. Ожидается ежегодный прирост в этом секторе на 21,3%.
Сектор рынка ДНК-диагностики патогенов в пище в настоящее время является самым скромным. В настоящее время на ДНК-диагностику микробного загрязнения производимых продуктов питания приходится менее 5% от общего количества методов, применяемых для тестирования загрязненности продуктов (культуральные методы и методы с использованием моноклональных антител). Однако методы ДНК-диагностики являются, по оценкам экспертов, более быстрыми, точными и информативными, что в ближайшее время, по-видимому, приведет к резкому возрастанию доли данного сектора рынка ДНК-диагностики.[11]
1.2. Полимеразная цепная реакция
ПЦР - это осуществляемая in vitro специфическая амплификация нуклеиновых кислот, инициируемая синтетическими олиго-нуклеотидными праймерами; основные ее этапы представлены на рис. 2. ПЦР-цикл состоит из тепловой денатурации ДНК, ее отжига с праймером и удлинения цепи (элонгации); смена этих этапов происходит в результате простого изменения температуры. Праймеры при этом ориентируются на матрице так, что число раундов репликации растет экспоненциально, соответственно увеличивается и число копий специфической нуклеотидной последовательности.
Применение молекулярных методов для целей клинической диагностики ограничивается их невысокой чувствительностью и длительностью анализа. Так, для выявления нуклеиновой кислоты-мишени методом гибридизации in situ с применением радиоактивно меченного зонда необходимо, чтобы мишень присутствовала в препарате в нескольких тысячах копий. Нередко число аномальных последовательностей в клиническом препарате диагностически-значимо, но меньше этой величины и гибридизация может дать ложноотрицательный результат. В отличие от этого ПЦР позволяет выявить уникальную нуклеотидную последовательность. Для этого в реакционную смесь добавляют в большом избытке специфичные для данной последовательности олигонуклеотидные праймеры («амплимеры»), которые образуют с ней комплекс, и проводят репликацию ДНК in vitro. Поскольку амплимеры гибридизуются с обеими цепями ДНК, то и нативная последовательность, и синтезируемые ПЦР-продукты могут служить матрицами в последующих раундах репликации, в результате чего число копий уникальной последовательности экспоненциально увеличивается (рис. 2). Благодаря этому последовательности, присутствующие в клиническом препарате в минимальном количестве (одна или несколько копий) и не поддающиеся обнаружению никакими другими методами, легко выявляются с помощью ПЦР. ПЦР позволяет найти всего одну аномальную последовательность на 100000-1000000 нормальных клеток.
Принцип полимеразной цепной реакции изображен на рис №1. Принцип комплиментарности и антипараллельности цепей ДНК представлен на рис №3.
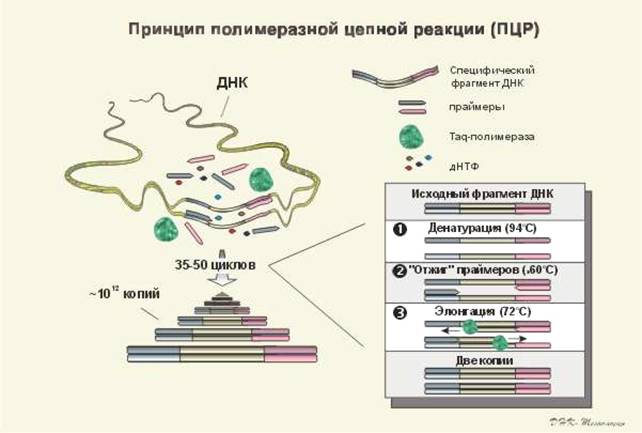
Рис №1.
Экспоненциальное увеличение числа копий молекулы-мишени не только обеспечивает высокую чувствительность метода, но и облегчает их выявление. Каждый раунд ПЦР занимает от 2 до 5 мин, и обычно для достижения необходимой чувствительности достаточно 25-50 раундов, т. е. 2-4 ч. Таким образом, весь анализ можно провести в течение одного дня по данным приведенным в [1], нам это удается за 3 ч. Кроме того, поскольку содержание ПЦР-продуктов достаточно велико, можно использовать неизотопные методы детекции. В табл.1 приведены данные, позволяющие сравнить метод ПЦР с другими молекулярно-генетическими методами исследования. Видно, что он обладает двумя важными преимуществами: высокой чувствительностью и непродолжительностью анализа.
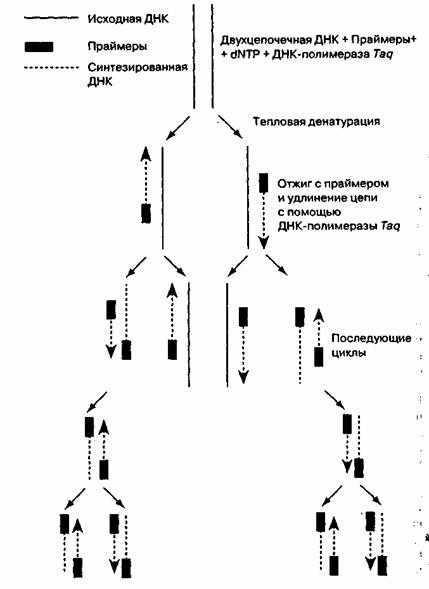
Рис. 2. Полимеразная цепная реакция. Праймеры подбирают таким образом, чтобы обеспечить специфическую амплификацию фрагмента ДНК определенного размера.
Таблица 1. Сравнение различных методов гибридизации
| ПЦР |
Блот-гибридизация по Саузерну |
Гибридизация in situ |
|
| Чувствительность |
1/100000 |
1/100 |
10 мишеней |
| Специфичность |
Высокая |
Высокая |
Высокая |
| Длительность анализа |
1 сут |
1 сут |
1 сут |
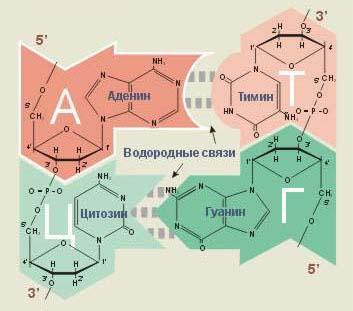
Рис. № 3. Принцип комплиментарности и антипараллельности цепей ДНК.
1.3. Амплификация РНК
Возможность использования РНК в качестве мишени для ПЦР существенно расширяет спектр применения этого метода. Например, геномы многих вирусов (гепатит С, вирус инфлюэнцы, пикорнавирусы и т.д.) представлены именно РНК. При этом в их жизненных циклах отсутствует промежуточная фаза превращения в ДНК. Для детекции РНК необходимо в первую очередь перевести ее в форму ДНК. Для этого используют обратную транскриптазу, которую выделяют из двух различных вирусов: avian myeloblastosis virus и Moloney murine leukemia virus. Использование этих ферментов связано с некоторыми трудностями. Прежде всего, они термолабильны и поэтому могут быть использованы при температуре не выше 42° С., Так как при такой температуре молекулы РНК легко образуют вторичные структуры, то эффективность реакции заметно снижается и по разным оценкам приблизительно равна 5%. Предпринимаются попытки обойти этот недостаток используя в качестве обратной транскриптазы термостабильную полимеразу, полученную из термофильного микроорганизма Thermus Thermophilus , проявляющего транскриптазную активность в присутствии Mn2+. Это единственный известный фермент, способный проявлять как полимеразную, так и транскриптазную активность.
Для проведения реакции обратной транскрипции в реакционной смеси также как и в ПЦР должны присутствовать праймеры в качестве затравки и смесь 4-х дНТФ, как строительный материал.
После проведения реакции обратной транскрипции, полученные молекулы кДНК могут служить мишенью для проведения ПЦР.
2.Общие положения PCR
Способы оптимизации условий, в которых протекает ПЦР рассмотрены в обзорах [4, 5]. Важную роль играют такие факторы как число циклов, временные и температурные характеристику процесса, концентрация амплимеров, ионов магния, дезоксинуксинуклеотидтрифосфатов, фермента. Стандартные реакционные смеси для ПЦР производит ряд коммерческих фирм (например, Perkin-Elmer Cetus). Сфера применения ПЦР все более расширяется, и в настоящее время описаны процедуры выявления различных последовательностей-мишеней, а для многих случаев в продаже имеются соответствующие наборы реактивов. В клинической практике желательно применять наборы для ПЦР-анализа, производимые коммерческими фирмами. Мы рассмотрим применение описанных ПЦР-методов в клинике и возможность их модификации с целью проведения рутинных клинических анализов.
2.1. Подбор праймеров
ПЦР-праймеры, или амплимеры, обычно имеют размер от 18 до 25 нуклеотидов. Их можно синтезировать с помощью автоматических синтезаторов ДНК, имеющихся в большинстве крупных исследовательских центров. Количества получаемых таким образом олигонуклеотидов (0,2—1 мкмолей) обычно достаточно для проведения нескольких сотен или тысяч ПЦР-реакций. Праймеры можно также приобрести у ряда коммерческих фирм (например, DuPont, Perkin—Elmer Cetus). Часто в 5’-концевой участок праймеров для упрощения клонирования ПЦР-амплифицированной ДНК вводят сайты узнавания для рестриктирующих эндо-нуклеаз.
Подбор праймеров - ключевое звено ПЦР, поскольку именно ими определяется возможность амплификации и выявления нужной последовательности, а также чрезвычайная гибкость метода. Простое варьирование праймеров позволяет выявлять многие патогенные микроорганизмы и генетические нарушения при минимальных изменениях в методике.[1]
Необходимо отметить, что праймеры могут отжигаться друг с другом, образуя праймер-димеры. И то, и другое приводит к значительному расходу праймеров на синтез побочных (неспецифических) продуктов реакции и, как следствие, значительно уменьшает чувствительность системы. Это затрудняет или делает невозможным чтение результатов реакции при проведении электрофореза.[22]
ПЦР-амплифицированные фрагменты ДНК имеют разный размер. Он определяется суммой размера праймеров и расстояния между их 3’-концами и в большинстве случаев лежит в диапазоне 100—300 нуклеотидов. Возможна амплификация (но обычно менее успешная) последовательностей-мишеней длиной более 1000 п.н. Скорость репликации с помощью ДНК-полимеразы Tag составляет 35—100 нуклеотидов в секунду.[1]
Из всего выше сказанного можно сформировать следующие требования к праймером:
1. Праймеры должны быть специфичны. Особое внимание уделяют 3’-концам праймеров, т.к. именно с них начинает достраивать комплиментарную цепь ДНК Taq-полимераза. Если их специфичность недостаточна, то, вероятно, что в пробирке с реакционной смесью будут происходить нежелательные процессы, а именно, синтез неспецифической ДНК (коротких или длинных фрагментов). Она видна на электрофорезе в виде тяжелых или легких дополнительных полос, иногда шмеров, выглядящих сплошным мазком на электрофорезе. Это мешает оценке результатов реакции, т.к. легко перепутать специфический продукт амплификации с синтезированной посторонней ДНК. Часть праймеров и дНТФ расходуется на синтез неспецифической ДНК, что приводит к значительной потере чувствительности.
2. Праймеры не должны образовывать димеры и петли, т.е. не должно образовываться устойчивых двойных цепей в результате отжига праймеров самих на себя или друг с другом.
3. Область отжига праймеров должна находиться вне зон мутаций, делеций или инсерций в пределах видовой или иной, взятой в качестве критерия при выборе праймеров, специфичности. При попадании на такую зону, отжига праймеров происходить не будет, и как следствие - ложноотрицательный результат. Иными словами, если предполагается разработать видоспецифическую тест-систему на какой-либо микроорганизм, выбирается именно та зона, которая удовлетворяет этим требованиям в пределах сиквенсов данного вида. И наоборот, такие же сиквенсы всех близкородственных видов должны иметь как можно более существенные отличия в последовательности ДНК
Таg-полимераза. В 1980 году полимеразная активность была открытая у некоторых форм термофильных бактерий. Таg-полимераза была выделена из бактерий вида Thermus aquaticus, способных расти при температуре 70-75°С. Молекулярный вес очищенного протеина 94 кД и оптимальная температура полимеразной активности 70-80°С. Активность фермента уменьшается, но полимераза не денатуруеться при 90°С, при снижении температуры до 70-80°С уровень активности восстанавливается.
Tag-полимераза имеет очень высокую скорость синтеза. При оптимальных условиях фермент может достраивать до 150 основ на секунду. При низкой температуре активность падает до 2 основ на секунду. Время полужизни Таg-полимерази при 95°С составляет 40 минут.[2]
В клинической практике, особенно при работе с недостаточно хорошо сохранившимися образцами тканей, часто приходится иметь дело с деградированной ДНК в этих случаях лучше ориентироваться на последовательности-мишени небольшого размера.
2.1.1. Консервативные последовательности-мишени
Чтобы ПЦР прошла успешно, должна произойти гибридизация амплимера с нужной последовательностью-мишенью. Если эта последовательность слегка различается у разных индивидуумов или у микроорганизмов из разных изолятов (т. е. имеет место полиморфизм), может произойти ее неполное спаривание с амплимером и нарушение нормальной амплификации, что приведет к получению ложноотрицательного результата. К счастью, у человека большая часть геномных последовательностей консервативна и не различается у разных индивидуумов, так что обычно для них можно использовать один набор «консервативных» (generic) амплимеров.
2.1.2. Полиморфные последовательности-мишени
В случае полиморфных мишеней, не полностью гомологичных выбранным праймерам, гибридизация и удлинение праймеров могут вообще не произойти. Наиболее критичным при этом является неправильное соответствие матрице 3’-концевых нуклеотидов амплимера. Показано, что небольшое нарушение гомологии между 3’-концом праймера и матрицей не сказывается на ходе реакции, если концентрация нуклеозидтрифосфатов находится на уровне, обычном для ПЦР. При низкой же концентрации нуклеозидтрифосфатов такое нарушение блокирует амплификацию. Таким образом, с помощью ПЦР можно различить аллели, различающиеся всего одним нуклеотидом.
Полиморфизм ДНК - весьма распространенное явление, поэтому очень важно знать, в каких случаях он имеет место и в какой степени. Предсказать полиморфизм у микроорганизмов невозможно, его можно выявить, только исследовав множество изолятов. Полиморфные локусы ДНК человека хорошо изучены и охарактеризованы и для их анализа можно подобрать оптимальные праймеры и гибридизационные зонды. Во многих случаях полиморфизм возникает в результате функционально значимых мутаций в половых или соматических клетках, и для выявления таких мутаций можно создать специальные праймеры и гибридизационные зонды. Анализ полиморфизма может использоваться также для идентификации личности (ПЦР-отпечаток пальцев [8]), на пример VNTR, STR полиморфизм [3].
Что касается микроорганизмов, то данных об их полиморфизме гораздо меньше, и результаты ПЦР-тестов могут оказаться ложноотрицательными. В этом случае полиморфизм может отражать аллельные различия между изолятами или возникать в результате нестабильности генома (например, у ретровирусов). В связи с ограниченностью данных о структуре генома микроорганизмов приходится определять специфичность набора праймеров и зондов экспериментально. Идеальный набор праймеров должен выявлять все изоляты исследуемого вида, но не близкородственные и различающиеся с медицинской точки зрения виды. Например, если разработанная комбинация праймеров и зондов специфична для одного из видов герпесвирусов, то она не должна «замечать» другие виды этого вируса, обеспечивая при этом амплификацию генетического материала всех изолятов исследуемого вида вируса. Поскольку даже у изолятов последовательности-мишени могут слегка различаться, для их идентификации используют два набора зондов и праймеров, специфичных к двум разным мишеням. Это гарантирует успех ПЦР-теста, поскольку одновременный полиморфизм по двум мишеням в пределах одного изоля-та маловероятен.
В работе [9] описана методика обнаружения вирусов папилломы человека (HPV), в которой для типирования используются как консервативные, так и полиморфные участки генома. При этом с помощью праймеров, специфичных в отношении консервативных участков, амплифицируют ДНК большого числа изолятов, а с помощью направленных на полиморфные последовательности типоспецифичных зондов различают изоляты. Можно также использовать разные наборы праймеров и зондов, специфичные к полиморфным участкам генома, и амплифицировать и типировать разные последовательности-мишени.
Для повышения чувствительности и специфичности можно использовать также «гнездовые» праймеры. При этом в роли матрицы выступает небольшая часть ДНК, амплифицированной в ходе первой ПЦР-реакции, а во второй ПЦР-реакции применяются праймеры, расположенные между «первыми» последовательностями-мишенями. Однако «гнездовые» праймеры не очень пригодны для рутинных клинических анализов, поскольку манипуляции с ПЦР-амплифицированной ДНК перед второй амплификацией повышают вероятность ее загрязнения.
2.2. Приборное обеспечение
Важным фактором воспроизводимости реакции амплификации является приборное обеспечение. Кроме надежности амплификатора и точности поддержания температур следует упомянуть о таком важном качестве прибора, как использование «активного регулирования», позволяющего добиваться достижения нужной температуры реакционной смеси внутри пробирки в значительно более короткие сроки, чем при обычном регулировании. Тем самым, сокращается время реакции (в 1,5-2 раза), увеличивается время сохранения активности Taq-полимеразы, что позволяет увеличить количество циклов амплификации, снизить риск неспецифического отжига праймеров, а следовательно, повысить чувствительность и специфичность реакции.
К приборам такого класса следует отнести аппараты фирмы Perkin-Elmer 2400 или 9600, HyBaid “TouchDown”, MJ Research PTC200. Среди отечественных амплификаторов активное регулирование реализовано в термоциклере MC-2 (“Терцик”), производимом нашей фирмой. При использовании приборов с активным регулированием следует учитывать тип ПЦР-пробирок и строго придерживаться рекомендаций фирм-изготовителей. Это объясняется тем, что при высоких скоростях изменения температуры минимальные отличия в конфигурации гнезд амплификатора и формы конуса ПЦР-пробирки могут сводить "на нет" преимущества активного регулирования и приводить к снижению чувствительности реакции из-за температурных погрешностей, связанных с ухудшением теплового контакта.
2.3. Число циклов
Чем больше число ПЦР-циклов, тем выше чувствительность метода. Однако максимальной чувствительности нельзя достичь только таким путем, необходимо оптимизировать и другие параметры, с тем чтобы повысить эффективность каждого цикла. Число циклов амплификации зависит также от содержания в образце нужной последовательности. Если ставится цель выявления уникальных генов, присутствующих в каждой клетке (например, при ПЦР-диагностике наследственных заболеваний), то достаточно 20—30 циклов, если же последовательность-мишень имеется только в части клеток исследуемого препарата (например, в случае вирусной инфекции или в опухолях), то число циклов может составлять 30—50.
2.4. Точность
Для проведения ПЦР-тестов обычно используют ДНК-полимеразу, выделенную из Thermus aquaticus. У этого фермента отсутствует корректирующая 3'-4’-5’-активность и частота ошибок при считывании достигает 1/10 000 нуклеотидов. Этой точности (хотя она и ниже, чем у многих других полимераз) достаточно для того, чтобы возникающие при репликации последовательности-мишени ошибки не создавали особых проблем.
2.5. Модифицированные методы
Классический способ постановки ПЦР, принципы которого были изложены выше, нашел свое развитие в некоторых модификациях, направленных на преодоление ограничений ПЦР и повышение эффективности прохождения реакции.
2.5.1. Постановка ПЦР с использованием “горячего старта”
Чтобы уменьшить риск образования неспецифических продуктов реакции амплификации, используют подход, получивший название “горячий старт” (“Hot-start”). Суть его состоит в предотвращении возможности начала реакции до момента достижения в пробирке условий, обеспечивающих специфический отжиг праймеров.
Дело в том, что в зависимости от ГЦ-состава и размера, праймеры имеют определенную температуру плавления (Tm). Если температура системы превышает Тm, праймер не в состоянии удерживаться на цепи ДНК и денатурирует. При соблюдении оптимальных условий, т.е. температуры отжига, близкой к температуре плавления, праймер образует двухцепочечную молекулу только при условии его полной комплементарности и, таким образом, обеспечивает специфичность реакции.
Существуют различные варианты реализации «горячего старта»:
1. Внесение в реакционную смесь Taq-полимеразы во время первого цикла после прогрева пробирки до температуры денатурации.
2. Разделение ингредиентов реакционной смеси парафиновой прослойкой на слои (в нижней части - праймеры, в верхней - Taq-полимераза и ДНК-мишени), которые смешиваются при расплавлении парафина (~65-750С) (рис. 4).
3. Использование моноклональных антител к Taq-полимеразе. Фермент, связанный моноклональными антителами, становится активным лишь после стадии первой денатурации, когда моноклональные антитела необратимо денатурируют и освобождают активные центры Taq-полимеразы.
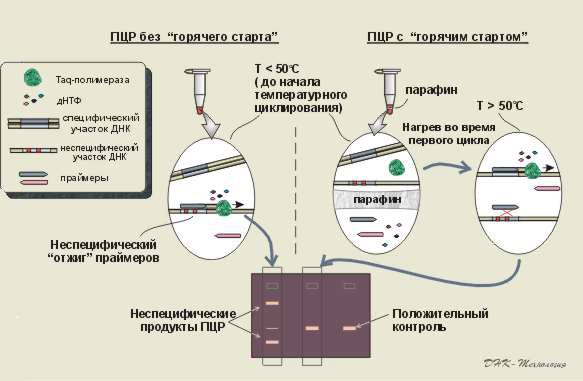
Рис № 4.
Во всех перечисленных случаях, даже если неспецифический отжиг произошел до начала температурного циклирования, элонгации не происходит, а при нагревании комплексы праймер-ДНК денатурируют, поэтому неспецифические продукты не образуются. В дальнейшем температура в пробирке не опускается ниже температуры плавления, что обеспечивает образование специфического продукта амплификации.
2.5.2. «Nested» ПЦР
Одним из способов повысить чувствительность реакции является применение метода «гнездной» (“nested”) амплификации. Суть его заключается в последовательном использовании двух пар праймеров – внешней и внутренней. После использования первой пары праймеров продукт амплификации переносят в другую пробирку с внутренней парой праймеров. Иногда вместо «гнездной» амплификации используют процесс реамплификации. В этом случае проводят дополнительный раунд амплификации, в котором используют прежнюю пару праймеров, а в качестве ДНК-мишени – продукт первой реакции амплификации.
Такая схема постановки амплификации более трудоемка и требует особенно тщательного обустройства лабораторных помещений, позволяющих гарантированно избегать контаминации продуктами реакции после использования внешней пары праймеров. К «гнездной» амплификации или реамплификации прибегают лишь в особых случаях, так как современные ПЦР-тест-системы позволяют добиваться тех же результатов иными средствами.
2.5.3. Полуколичественный анализ.
Для более точной оценки количества ДНК-мишени в реакционной смеси предпринимаются специальные подходы, известные под общим названием полуколичественного ПЦР-анализа. Приставка полу- имеет принципиальное значение из-за условной точности результатов такого анализа.
К таким подходам следует отнести использование специального приборного обеспечения, а также введение в реакционную смесь внутренних контролей с разной концентрацией.
К приборному обеспечению, позволяющему получать полуколичественные результаты ПЦР анализа, следует отнести последнюю модель амплификатора фирмы Perkin Elmer, которая позволяет следить за кинетикой накопления продуктов амплификации. Это достигается красивым решением на стыке физики и биологии. В реакционную смесь вводят гибридизационные зонды, в состав которых входят меченные особыми реактивами нуклеотиды, способные флуоресцировать лишь в свободном состоянии. На стадии отжига происходит гибридизация зондов с внутренними участками ампликонов. В процессе элонгации Taq-полимераза разрушает зонды за счет своей экзонуклеазной активности. Это приводит к попаданию меченых нуклеотидов в раствор, где они начинают флуоресцировать. Интенсивность флуоресценции фиксируется специальным детектором и соответствует количеству продуктов амплификации. Если гибридизация не проходит, то зонд остается целым, а нуклеотиды, входящие в его состав, не флуоресцируют.
При исследовании образцов каждая серия экспериментов сопровождается постановкой амплификации с контролями (несколько 10-кратных разведений ДНК). Сравнение кинетики образования свободных нуклеотидов в реакционных смесях в экспериментальном образце и контрольных пробирках позволяет полуколичественно оценить концентрацию ДНК в образце в диапазоне разведений контрольных препаратов ДНК.
Более простым, но менее надежным вариантом оценки количества специфической ДНК является сравнение конечных результатов одновременной амплификации в одной пробирке образца и контрольной ДНК (внутреннего контроля). При этом ставят несколько реакций с различными разведениями внутреннего контроля. Детекцию проводят после гибридизации с ДНК-зондами, или методом электрофореза (в последнем случае продукты амплификации исследуемой ДНК и внутреннего контроля должны отличаться по размеру).
При всей привлекательности полуколичественного анализа следует отметить некоторые его существенные недостатки.
1. Для полуколичественного анализа допускается использование только препаратов с высокой степенью очистки ДНК. Важно отметить, что существуют примеси, которые могут по-разному влиять на эффективность амплификации исследуемой и контрольной ДНК. При использовании рутинных методов выделения ДНК в большинстве случаев не представляется возможным заранее предсказать количество и состав примесей в клинических образцах.
2. Из-за существования эффекта «плато», а также конкуренции за компоненты реакции при использовании «внутренних контролей» для оценки количества ДНК необходима постановка целого ряда реакций амплификации, что делает этот метод трудоемким и дорогостоящим.
3. Известно, что в некоторых случаях возможны потери ДНК на стадии выделения, что может существенно исказить значение реального количества ДНК в образце.
4. Даже если предположить, что все компоненты, входящие в состав ПЦР-набора, идеально стандартизованы, нет гарантии, что часть ДНК не деградировала в процессе хранения образца.
5. Кроме того, точное знание количества возбудителей в конкретном клиническом образце далеко не всегда может дать полное представление об инфекционном процессе, например, из-за различной локализации и вирулентности микроорганизмов.
Таким образом, существующие варианты полуколичественного анализа пока еще не имеют большой практической ценности для рутинного клинического тестирования и могут быть использованы только в некоторых особо сложных случаях или для научных целей.[26]
3. Работа с образцами тканей
ПЦР-анализ состоит из трех стадий изображенных на рис. 5.
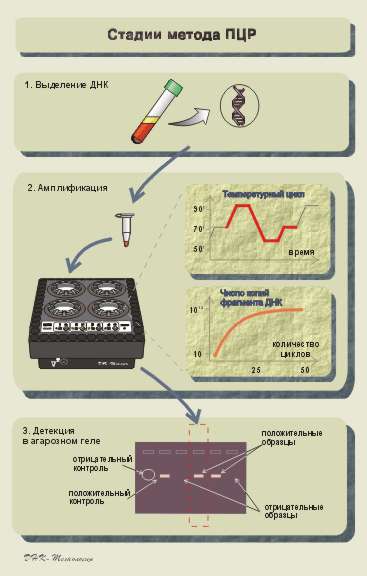
Рис. 5. Три этапа ПЦР-анализа
3.1. Подготовка пробы биологического материала.
Для выделения ДНК используют различные методики в зависимости от поставленных задач. Их суть заключается в экстракции (извлечении) ДНК из биопрепарата и удалении или нейтрализации посторонних примесей для получения препарата ДНК с чистотой, пригодной для постановки ПЦР.
Иногда бывает достаточно прокипятить образец в течение 5-10 мин., однако в большинстве случаев требуются более сложные методы.
Стандартной и ставшей уже классической считается методика получения чистого препарата ДНК, описанная Мармуром описана в п.3.1.1. Она включает в себя ферментативный протеолиз с последующей депротеинизацией и переосаждением ДНК спиртом. Этот метод позволяет получить чистый препарат ДНК. Однако он довольно трудоемок и предполагает работу с такими агрессивными и имеющими резкий запах веществами, как фенол и хлороформ.
Одним из популярных в настоящее время является метод выделения ДНК, предложенный Boom с соавторами. Этот метод основан на использовании для лизиса клеток сильного хаотропного агента - гуанидина тиоционата (GuSCN), и последующей сорбции ДНК на носителе (стеклянные бусы, диатомовая земля, стеклянное «молоко» и.т.д.). После процесса очистки в пробе остается ДНК, сорбированная на носителе, с которого она легко снимается с помощью элюирующего буфера. Метод удобен, технологичен и пригоден для подготовки образца к амплификации. Однако возможны потери ДНК вследствие необратимой сорбции на носителе, а также в процессе многочисленных очисток. Особенно большое значение это имеет при работе с небольшими количествами ДНК в образце. Кроме того, даже следовые количества GuSCN могут ингибировать ПЦР. Поэтому при использовании этого метода очень важен правильный выбор сорбента и тщательное соблюдение технологических нюансов. Следует отметить, что из-за большого количества стадий добавления и удаления растворов при работе с образцом требуется аккуратность, т.к. возможна перекрестная контаминация между пробами образующейся аэрозолью ДНК.
Другая группа методов пробоподготовки основана на использовании ионообменников типа Chilex, которые, в отличие от стекла, сорбируют не ДНК, а наоборот, примеси, мешающие реакции. Как правило, эта технология включает две стадии: кипячение образца и сорбция примесей на ионообменнике. Метод чрезвычайно привлекателен простотой исполнения. В большинстве случаев он пригоден для работы с клиническим материалом. К сожалению, иногда встречаются образцы с такими примесями, которые невозможно удалить с помощью ионообменников. Кроме того, некоторые микроорганизмы не поддаются разрушению простым кипячением. В этих случаях необходимо введение дополнительных стадий обработки образца.
При массовом скрининге, когда важно получить статистические данные, возможно использование простых методов с применением детергентов или обработки биологического материала щелочами с последующей их нейтрализацией. В то же время, использование подобных методов пробоподготовки для клинической диагностики может приводить к ложноотрицательным результатам, вследствие использования в реакционной смеси некачественного препарата ДНК.
Таким образом, к выбору метода пробоподготовки следует относиться с пониманием целей проведения предполагаемых анализов.
Образцы тканей, получаемые при хирургических операциях, обычно фиксируют формалином и заливают в парафин. Такие фиксированные препараты могут использоваться для проведения ПЦР методика выделения описана в п. 3.1.2.
3.1.1. Нативные ткани
Оптимальной матрицей для ПЦР является ДНК, выделенная из нативных тканей обычными методами. Эффективность амплификации такой ДНК обычно выше, чем ДНК, выделенной из фиксированных тканей, поэтому сравнивать результаты, полученные в этих двух случаях, следует с осторожностью.
Метод выделения ДНК из клеток крови с протеиназой К
1. К 1 объему крови (1-5 мл) перемешивать добавить 4-6 объема сахарозного буфера (приготовленного согласно табл. 3) охладить до 4 0С.
2. Центрифугируем 10 мин при Fr (критерий Фруда)=1700 (3000 об.мин).
3. Полученный осадок ресуспендировать палочкой до получения гомогенной смеси.
4. В зависимости от количества осадка добавляем следующие реагенты (Табл №2). Реагенты надо добавлять в следующей последовательности: протеиназный буфер (приготовленного согласно табл. 4) тщательно перемешать, SDS (тщательно перемешать, наблюдаем изменение окраски и появление вязкости), протеиназа К (тщательно перемешать). Для каждой манипуляции при последующей работе используем новый носик для самплера.
Табл № 2.
| Осадок |
Протеиназный буфер (таблица 3) мл |
SDS Мкл |
Протеиназа К Мкл |
| Слабо виден |
470 |
25 |
5 |
| Нормально |
940 |
50 |
10 |
| Много |
1880 |
100 |
20 |
5. Поставить в термостат на 16 ч. при t=37 0С или на 2ч. при 55 0С.
Экстракция и очистка
6. Добавить равный объем фенола, перемешиваем 10 мин.
7. Центрифугировать 5-10 мин при Fr=3000.
8. Отбираем супернантант и переносим в новую пробирку.
9. К супернантанту добавить равный объем фенола/хлороформ в соотношении 1:1, перемешиваем 10 мин. Для экстракции белков.
10. Центрифугируем 5-10 мин при Fr=3000.
11. Отбираем супернатант и переносим в новую пробирку.
12. Добавить равный объем хлороформа, перемешиваем в течении 10 мин.
13. Выполнять пп. 11,12. Пока не очистится интерфаза (2-3 раза).
14. Отбираем супернатант в конусный цилиндр.
15. Добавить 2,5 объема Et-OH (96%) при Т=-200С.
16. Формируется “медуза” которую наматываю на стеклянную палочку, проводим промывку 70% этанолом.
17. Проводим сушку на протяжении 5..10 мин.
18. Растворить ДНК в деионизированной воде или ТЕ.
Для растворение ставят в термостат на несколько часов.
Табл.№3. Состав сахарозного буфера.
| Реактив |
Химическачя формула |
Концентрация стока |
Концентрация М |
V (на 500мл) |
| Сахароза |
Чистое в-во |
0,32 |
55г. |
|
| Тритон X-100 |
|
Чистое в-во |
1%(обемный) |
5мл |
| TRIS-HCl |
0,1М |
0,001 |
5мл |
|
| MgCl2 |
4,82М |
0,0482 |
0,5мл |
|
Вода |
55,55 М |
55,18 |
До 500мл |
Табл.№4. Состав протеиназного буфера
TRIS-HCl |
1М |
0,01 |
100мкл |
EDTA |
0,5М |
10-5 |
0,2мкл |
NaCl |
5М |
10-4 |
0,2мкл |
Вода |
До 10мл |
3.1.2. Фиксированные ткани
3.1.2.1. Фиксирующие вещества
Ткани лучше всего фиксировать сразу после их получения, однако этот момент не является ключевым. Можно проводить амт лификацию ДНК и в том случае, если ткани получены через семь (а возможно, и более) дней после смерти. Отличной матрицей для ПЦР является ДНК тканей, фиксированных в 10% забуференном формалине и залитых в парафиновые блоки. Более того; при осторожном выделении ДНК из таких фиксированных тканей можно получить молекулы, имеющие достаточную длину для проведения Саузерн-гибридизации. Вполне пригодным для ПЦР материалом являются ткани, фиксированные в этаноле или ацетоне и залитые в парафин. Несколько худшие результаты получаются при фиксации тканей фиксаторами Кларка и Замбони, параформальдегидом, формалином / этанолом уксусной кислотой, метакарном и совсем непригодны фиксаторы Зенкера, Карнуа, Буэна, В-5. Более полную информацию о способах фиксации, пригодных для дальнейшего проведения ПЦР, можно найти в работе [14]. Обработка препаратов кислыми растворами, декальцифицирующими костную ткань, обычно приводит к деградации ДНК и тем самым препятствует амплификации.
3.1.2.2. Длительность фиксации
Длительная (более 3 сут) фиксация ткани в 10% забуференном формалине недопустима. Оптимальное время фиксации перед ее дальнейшей обработкой составляет от 1 до 24 ч. Следует учесть также, что в большинстве случаев при такой обработке происходит дополнительная фиксация в 10% формалине.
Под действием формальдегида в ДНК образуются шиффовы основания; в водных растворах эта реакция обратима. При увеличении времени фиксации начинаются другие, необратимые процессы. После заливки ткани в парафин стабильность ДНК повышается, но медленная ее деградация все-таки происходит, и в очень старых (более 10 лет) парафиновых блоках эффективность амплификации может быть меньше, чем в свежеприготовленных. Тем не менее в большистве случаев с помощью ПЦР удается анализировать и 30-летние парафинизированные препараты, а иногда даже археологические образцы.
3.1.2.3. Изготовление срезов
Срез ткани помещают в стерильную микроцентрифужную пробирку на 1,5 мл так, чтобы исключить загрязнение его посторонней тканью. Толщина среза составляет 5—10 мкм, площадь варьирует от 1 мм2 до 1-3 см2. Работать можно как в резиновых перчатках, так и без них. Мы в своей лаборатории перчатки не используем, поскольку в них очень трудно выполнять тонкие операции, а просто тщательно моем руки. Для манипуляций со срезами можно также использовать фильтровальную бумагу. В препарат может попасть незначительное количество клеток с пальцев исследователя, что необходимо учитывать при интерпретации результатов. Нежелательно, чтобы изготовлением срезов занимался человек с кожными заболеваниями (например, паракератозом), бородавками (если нужно выявить вирусы папилломы) и т. д. Более серьезную проблему составляет перекрестное загрязнение образцов тканей. Чтобы избежать этого, необходимо автоклавировать (в течение примерно 20 мин) и тщательно маркировать микроцентрифужные пробирки. Микротом нужно очистить от следов парафина и тканей, а нож перед получением очередного среза осторожно протереть фильтровальной бумагой. Менять ножи или протирать их кислотами или ксилолом обычно не требуется, достаточная очистка происходит при подравнивании блока перед получением среза. Для проверки, чистоты препаратов нужно поочередно делать срезы тканей, представляющих собой положительный и отрицательный контроли.
Лишний парафин с блоков обычно не удаляют. Микроцентрифужные пробирки на 1,5 мл, куда помещают срезы, должны быть сухими, поскольку вода мешает депарафинированию. Если срезы очень маленькие, их лучше сразу поместить в пробирку, где будет проводиться ПЦР (микроцентрифужная пробирка на 0,5 мл). Полученные срезы можно хранить в течение месяцев при комнатной температуре.
Большинство тканей являются хорошими субстратами для ПЦР. Иногда возникают проблемы при анализе таких тканей, как мозг или селезенка, но особенно трудно работать с препаратами, сильно загрязненными кровью, и с некротизированными тканями.
3.1.2.4. Выделение ДНК из срезов
Парафин из срезов удаляют обычными методами (протокол 1). В зависимости от размера среза для выделения ДНК используют два основных метода, которые описаны в протоколах 2 (выделение ДНК из небольших количеств ткани) и 3 (выделение ДНК из срезов большого размера). В табл. 5 даны рекомендации по выбору метода выделения.
Протокол 1. Депарафинирование срезов
Срезы можно депарафинировать с помощью ксилола или октана.
1. В микроцентрифужную пробирку на 1,5 мл, куда помещен срез, добавляют примерно 1 мл выбранного растворителя, встряхивают и оставляют при комнатной температуре примерно на 2 мин. Парафин при этом должен полностью раствориться.
2. Собирают ткань центрифугированием при 12 000 g в течение 5 мин.
3. Сливают супернатант (осадок при этом может быть едва заметен).
4.Добавляют в пробирку 1 мл 95-100% этанола и встряхивают (ткань при этом должна приобрести беловатый цвет).
5. Центрифугируют при 12 000 g в течение 5 мин и сливают супернатант.
6. Этапы 4 и 5 повторяют еще раз (необходимо дважды промыть ткань этанолом).
7. Высушивают ткань в эксикаторе или вакуумном концентраторе в течение 30 или более минут.
8. Высушенную ткань можно хранить при комнатной температуре в течение нескольких недель.
Таблица 5. Рекомендации по выбору метода выделения ДНК
| Площадь среза |
Число срезов |
Метод выделения |
| < 1 мм2 (очень маленькие биоптаты или клеточные блоки,полученные из тонкоигольных аспиратов) |
1-3 |
Протокол 2 |
| 1-2 мм2 (маленькие биоптаты) |
1-3 |
Протоколы 2 и 3 |
| > 2 мм2 (большинство тканей) |
1 |
Протокол 3 |
Протокол 2. Выделение ДНК из небольших срезов (см. табл. 5)
При работе с очень небольшими срезами амплификацию необходимо провести сразу после депарафинирования.
1. Добавляют 50 мкл стерильной дистиллированной воды к высушенному препарату, подготовленному так, как это описано в протоколе 1.
2. Микроцентрифужную пробирку плотно закрывают и на 7 мин помещают в кипящую водяную баню, затем охлаждают ее во льду.
3. Добавляют 50 мкл амплификационной смеси и измельчают ткань кончиком микропипетки, с тем чтобы обеспечить доступ к ДНК реактивов, входящих в состав смеси. Остатки ткани в микроцентрифужной пробирке при этом остаются видимыми.
4. Проводят амплификацию в термоциклере (см. протокол .4).
Протокол 3. Выделение ДНК из больших срезов (см. табл. 2)
Материалы
• Буфер для выделения: 100 мМ трис-НС1, 1 мМ ЭДТА, рН 8,0
• Протеиназа К: 20 мг/мл в стерильной дистиллированной воде
Методика
1.Добавляют 50 мкл буфера для выделения к высушенному препарату, подготовленному так, как это описано в протоколе 15.1.
2. Добавляют 1 мкл раствора протеиназы К.
3. Измельчают ткань кончиком микропипетки, с тем чтобы обеспечить доступ реактивов к ткани, и инкубируют ее при 37 °С в течение ночи или при 50 °С в течение 3 ч.
4. Встряхивают образец в течение 30 с на вортексе и кипятят его для инактивации протеиназы К в течение 7 мин.
5. Остатки ткани собирают центрифугированием при 12 000 g в течение 5 мин и отбирают супернатант, который в дальнейшем используют для проведения ПЦР.
3.2. Проведение ПЦР
Если образец ткани очень маленький, лучше амплифицировать всю полученную из него ДНК (см. протокол 2). В случае срезов большого размера для амплификации достаточно 1/10-1/50 материала. В некоторых случаях амплификация ингибируется самой выделенной ДНК (или какими-то другими компонентами, присутствующими в буфере для выделения), и лучшие результаты получаются при меньшем количестве материала. Количество выделенной ДНК и степень ее деградации можно определить, проведя электрофорез 5-10 мкл раствора ДНК, полученной в соответствии с протоколом 3, в 0,7% агарозном геле, окрашенном бромистым этидием.
Очень важным при анализе фиксированных тканей является размер последовательности-мишени. Обычно небольшие фрагменты детектировать проще. Лучше, чтобы их размер не превышал 200 п.н., а оптимальной является длина < 100 п.н., в этом случае амплификация практически всегда проходит успешно.
В ходе ПЦР образцы инкубируют при трех разных температурах, соответствующих трем этапам цикла: денатурации мишени (90-95 °С), отжигу (40-60 °С) и удлинению праймера (72 °С). При проведении амплификации вручную оборудование может быть очень простым: несколько водяных бань с разной температурой. В последнее время широко используют автоматические приборы с микропроцессорным управлением; их выпускают такие фирмы, как Perkin—Elmer Cetus Instruments, Techne, Koch-Light.
В протоколе 4 дано описание типичной ПЦР-реакции. Исходную реакционную смесь, приготовленную так, как это описано в п. 1, разливают по пробиркам, в которых будет проводиться амплификация, и добавляют ДНК-мишень в последнюю очередь. Объем реакционной смеси не очень критичен (достаточно точности ~ 10%), так что корректировать состав реакционной смеси в зависимости от разного количества добавленной ДНК-матрицы не нужно. Детали процесса амплификации, приведенные в п. 4, следует рассматривать как ориентировочные, но температура денатурации 94-95 °С приемлема для всех мишеней. Более длительный начальный прогрев (5 мин при 95 °С) обеспечивает полную денатурацию всех молекул ДНК в образце, и последующая денатурация в каждом цикле амплификации будет занимать меньше времени (45-60 с). Частой причиной неудачи является недостаточное нагревание образцов на этапе денатурации. Температура отжига определяет жесткость условий гибридизации праймеров с мишенью и, следовательно, специфичность амплификации; она зависит от длины праймеров и их GC-содержания. Эту температуру можно оценить по формуле
4 °С • (G + С) + 2 °С • (А + Т) - 3.
На практике температуру отжига необходимо подбирать опытным путем, используя положительные и отрицательные контроли; она находится в диапазоне 37-65 °С.
Специфичность ПЦР приведена на рис 6.
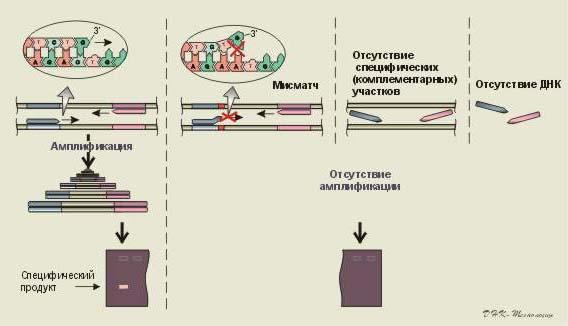
Рис 6.Специфичность ПЦР
Температура на этапе элонгации должна составлять 72 °С, именно при таких условиях ДНК-полимераза Tag обладает оптимальной активностью. Необходимое время элонгации определяется размером продуктов амплификации; считается, что копирование каждой 1000 п.н. занимает 1 мин. Таким образом, в случае архивных препаратов с размером мишени 100-200 п.н. время элонгации обычно составляет 40—60 с.
Результатом циклического синтеза является экспоненциальное увеличение количества специфического фрагмента ДНК см рис 7, которое можно описать формулой
А = М* (2n - n-1) ~ 2n ,
где А - количество специфических (ограниченных праймерами) продуктов реакции амплификации;
М – начальное количество ДНК-мишеней;
n - число циклов амплификации.
Фактически же значение эффективности отдельных циклов амплификации составляет, по некоторым данным, 78-97%. В случае присутствия в пробе ингибиторов реакции это значение может быть намного меньше, поэтому фактическое количество специфических продуктов амплификации лучше описывает уравнение
А = М* (1+Е)n,
где Е – значение эффективности реакции.
Следует заметить, что в процессе амплификации на исходной цепи синтезируются и длинные фрагменты, однако их накопление происходит лишь в арифметической прогрессии по формуле
К = М* n,
где К – количество длинных продуктов амплификации;
n - число циклов.
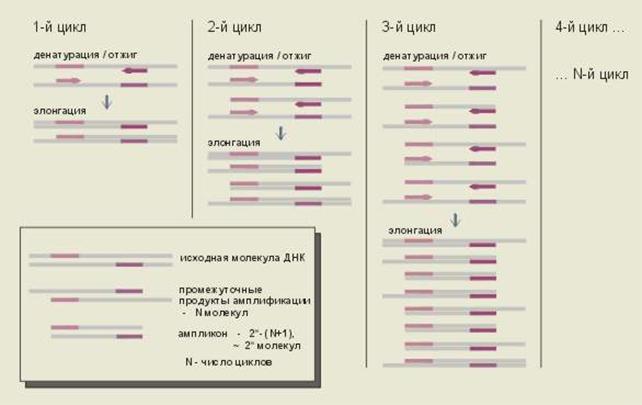
Рис. № 7. Удвоение числа синтезированных копий фрагмента ДНК на протяжении каждого из циклов ПЦР.
Таким образом, специфические фрагменты, ограниченные на концах праймерами, впервые появляются в конце второго цикла, накапливаются в геометрической прогрессии и очень скоро начинают доминировать среди продуктов амплификации.
Следует заметить, что процесс накопления специфических продуктов амплификации по геометрической прогрессии идет лишь ограниченное время, а затем его эффективность критически падает. Это связано с так называемым эффектом «плато».
Термин эффект “плато” используют для описания процесса накопления продуктов ПЦР на последних циклах амплификации, когда количество ампликонов достигает 0,3-1 пмолей.
В зависимости от условий и количества циклов реакции амплификации, на момент достижения эффекта “плато” влияют утилизация субстратов (дНТФ и праймеров), стабильность реактантов (дНТФ и фермента), количество ингибиторов, включая пирофосфаты и ДНК-дуплексы, конкуренция за реактанты неспецифическими продуктами или праймер-димерами, концентрация специфического продукта и неполная денатурация при высокой концентрации продуктов амплификации.
Чем меньше начальная концентрация ДНК-мишени, тем выше риск выхода реакции на “плато”. Этот момент может наступить до того, как количество специфических продуктов амплификации будет достаточно, чтобы их можно было проанализировать. Избежать этого позволяют лишь хорошо оптимизированные тест-системы.[16]
Протокол 4. Полимеразная цепная реакция
Материалы
• 10 х буфер: 100 мМ трис-HCl, рН 8,3, 500 мМ КС1, 25 мМ MgCl2
• 10x dNTP: пo2мM dATP, dCTP, dGTP, dTTP
• Праймеры: 20-100 пмоль каждого праймера на реакцию
• Tag-полимераза
• ДНК, выделенная в соответствии с протоколами 2 или 3
• Минеральное масло
Методика
1. Готовят смесь следующего состава:
• 10 х буфер 10 мкл
• l0 x dNTP 10 мкл
• Праймеры до конечной концентрации 0,2-1 мкМ
• Taq-полимераза 0,4 мкл (2 ЕД)
Если амплификация ингибируется ДНК-матрицей, то количество ДНК-полимеразы Tag можно увеличить (до 4 и более единиц)
2. Добавляют ДНК-матрицу и доводят объем реакционной смеси дистиллированной водой до 100 мкл.
3. На водную фазу наслаивают 1—2 капли минерального масла.
4. Помещают пробирки в прибор для амплификации, запрограммированный на определенный режим работы. Типичный ПЦР-цикл может быть таким: начальный прогрев при 94 °С в течение 5 мин; 20-50 циклов, состоящих из этапов денатурации (95 °С в течение 45 с), отжига (50 °С в течение 45 с), элонгации (72 °С в течение 60 с).
Протокол 5. Полимеразная цепная реакция
Материалы
• 10 х Неполный амплификационный буфер (НАБ):
Трис-HCl 1М 3,35млл, MgCl2 4,82М 15,56млл, 2-меркаптоэтанол 39 мкл,
EDTA 0.1M 35мкл, (NH4)2SO4 0.1095г., вода 1,49л.
• MgCl2
• 10x dNTP: пo2,5млM dATP, dCTP, dGTP, dTTP
• Праймеры: 20-100 пмоль каждого праймера на реакцию
• Бычий сыворотчатый альбумин (БСА): 0.17млг альбумина, вода 1мл
• Tag-полимераза 10 U/мкл
• ДНК, выделенная в соответствии с методом выделения ДНК из клеток крови с протеиназой К
• Праймеры 10мкМ
• Вода деионизированная
• Минеральное масло
Методика
1. Готовят смесь следующего состава:
| Реагент |
Начальная концентрация |
Конечная концентрация |
Обем |
| НАБ |
X10 |
X1 |
2,5 мкл |
| MgCl2 |
1 мкл |
||
| dNTP |
2.5 млМ |
0,2млМ |
2 мкл |
| БСА |
X25 |
X1 |
1мкл |
| Taq-полимераза |
10U/мл |
0,04U/мл |
100 нл |
| Праймеры |
1 о.е. |
0,06 о.е. |
1,5 мкл |
| Вода |
до 22 мкл |
2. Добавляют 3мкл ДНК-матрицу
3. На водную фазу наслаивают 1 каплю минерального масла.
4. Помещают пробирки в прибор для амплификации (амплификатор), запрограммированный на определенный режим работы. Типичный ПЦР-цикл может быть таким: начальный прогрев при 94 °С в течение 3 мин; 30 циклов, состоящих из этапов денатурации (94 °С в течение 40 с), отжига (55 °С в течение 60 с), элонгации (71 °С в течение 60 с). В случае образования неспецифичного продукта используют прогресивно-модифицированый цикл состояций из 4 програм. Температурный цикл PCR получения продукта используемого для выявления мутаций G542X представлен в табл. 6.
Табл 6.
| № программы |
№ шага |
Температура °С |
Время выдерживания с |
Количество циклов |
| 1 |
1 |
94 |
240 |
1 |
| 2 |
1 |
94 |
40 |
3 |
| 2 |
2 |
57 |
40 |
|
| 2 |
3 |
71 |
40 |
|
| 3 |
1 |
94 |
40 |
3 |
| 3 |
2 |
56 |
40 |
|
| 3 |
3 |
71 |
40 |
|
| 4 |
1 |
94 |
60 |
24 |
| 4 |
2 |
55 |
60 |
|
| 4 |
3 |
71 |
60 |
3.3. Спинномозговая жидкость (СМЖ)
Небольшой объем доступной для анализа спинномозговой жидкости не позволяет выделить из нее ДНК в количестве, достаточном для применения обычных молекулярно-генетических методов. Но, по нашим данным и данным других исследователей [18, 19], не содержащий крови препарат СМЖ можно исследовать, прокипятив его сразу после получения от больного. Такой препарат объемом до 50 мкл добавляют в реакционную смесь объемом 100 мкл. Соответствующая процедура описана в протоколе 6.
Протокол 6. ПЦР-анализ клеток СМЖ
С помощью ПЦР можно выявлять в СМЖ внеклеточные микроорганизмы (предварительно осадив их центрифугированием).
1. Определяют число присутствующих в образце лейкоцитов (например, при концентрации 10 клеток/мкл). Максимальное число грубо очищенных клеток, которое можно амплифицировать, равно примерно 100 000.
2. 1 мл СМЖ разливают в стерильные пробирки на 0,5 мл, где будет проводиться ПЦР:
| Объем СМЖ, мкл |
Объем воды, мкл |
Число клеток (при концентрации 10 клеток/мкл) |
| 10 |
40 |
100 |
| 50 |
0 |
500 |
| 200 |
0 |
2000 |
| 750 |
0 |
7500 |
3. Для разрушения клеток кипятят пробы в течение 5-10 мин.
4. Добавляют реакционную смесь для проведения ПЦР и осуществляют амплификацию в соответствии с тем, как это описано в протоколе 4.
3.4. Ткани, замороженные в среде, обеспечивающей оптимальные условия резания при данной температуре (ОСТ)
Часто образцы тканей замораживают в специальных средах (таких, как ОСТ), облегчающих получение срезов. Имея всего один замороженный срез толщиной 10 мкм, можно легко и быстро провести ПЦР, сохранив большую часть образца для дальнейших исследований (см. протокол 7). Эффективность ПЦР, в которой матрицей служит ДНК из замороженных срезов, обычно выше, чем для ДНК из фиксированных срезов.
Протокол 7. Обработка криостатных срезов
1. В стерильную пробирку помещают один криостатный срез толщиной 10 мкм.
2. Добавляют немного 0,9% (в/о) NaCl, быстро перемешивают, собирают ткань центрифугированием при 12 000 g в течение 5 мин и удаляют по возможности весь супернатант.
3. Фиксируют ткань в течение 5—10 мин в 95% этаноле.
4. Осаждают ткань центрифугированием при 12 000 g в течение 5 мин и высушивают ее.
5. Выделяют ДНК таким же образом, как из залитых в парафин тканей (см. протокол 3), и проводят амплификацию (см. протокол 4).
3.5. Архивные препараты
Чтобы амплифицировать ДНК из окрашенных архивных препаратов или из мазков, окрашенных по методу Папаниколау, препараты соскребают с предметных стекол и обрабатывают их так же, как высушенную депарафинированную ткань (протокол 4). При этом образуется большое число фрагментов ткани, что существенно повышает вероятность перекрестного загрязнения препаратов и требует особой аккуратности в работе.
4. Анализ ПЦР-амплифицированной ДНК
Для анализа ПЦР-амплифицированной ДНК используют разные методы. Мы рассмотрим три наиболее простых: гель-электрофорез, дот-блот-гибридизацию и блот-гибридизацию по Саузерну. С их помощью можно анализировать большинство ПЦР-продуктов, но абсолютно точные результаты можно получить только секвенированием.
4.1. Подготовка ПЦР-продуктов
Прежде всего необходимо удалить покрывавшее реакционную смесь минеральное масло. Для этого в пробирку для ПЦР добавляют несколько капель хлороформа, пробирку встряхивают и центрифугируют при 12 000 g в течение 1 мин, чтобы отделить водную фазу, содержащую ПЦР-продукты. Амплифицированную ; ДНК можно хранить с хлороформом при 4 °С в течение нескольких недель. Для последующего анализа обычно берут от 1/10 до 1/5 объема реакционной смеси.
При использивании ДНК выделения из клеток крови подготавительные работы непроводятся.
4.2. Гель-электрофорез
Электрофорез в агарозном геле (произведенный согласно инструкции 1) позволяет легко, без применения радиоизотопов, обнаружить амплифицированную ДНК и определить ее размер. Остановимся на некоторых ее особенностях применительно к анализу ПЦР-амплифицированной ДНК.
10—20 мкл амплифицированной ДНК разделяют в 2% агарозном геле с добавлением специального красителя ДНК, например, бромистого этидия вместе со стандартными фрагментами размером 50-1000 п.н. При заливке с помощью гребенок в геле формируют специальные лунки, в которые в дальнейшем вносят продукты амплификации. При использивании ДНК выделения из клеток крови используют 5 мкл амплифицированной ДНК смешивают с 3 мкл красителя на парафилме и наносят в лунку.
Пластину геля помещают в аппарат для горизонтального гель-электрофореза и подключают источник постоянного напряжения. Отрицательно заряженная ДНК начинает двигаться в геле от минуса к плюсу. При этом более короткие молекулы ДНК движутся быстрее, чем длинные. На скорость движения ДНК в геле влияет концентрация агарозы, напряженность электрического поля, температура, состав электрофорезного буфера и, в меньшей степени, ГЦ-состав ДНК. Все молекулы одного размера движутся с одинаковой скоростью. Краситель встраивается (интеркалирует) плоскостными группами в молекулы ДНК. После окончания электрофореза, продолжающегося от 10 мин до 1 часа, гель помещают на фильтр трансиллюминатора, излучающего свет в ультрафиолетовом диапазоне (254 - 310 нм). Энергия ультрафиолета, поглощаемая ДНК в области 260 нм, передается на краситель, заставляя его флуоресцировать в оранжево-красной области видимого спектра (590 нм).
Яркость полос продуктов амплификации может быть различной. Поэтому часто в ПЦР-лабораториях принято оценивать результат по трех- четырех или пяти- балльной системе. Однако, как уже отмечалось ранее, это нельзя связывать с начальным количеством ДНК-мишени в образце. Часто уменьшение яркости свечения полос связано со снижением эффективности амплификации под влиянием ингибиторов или других факторов.
Электрофорез проводят при высоком напряжении (10-15 В/см), поскольку образующиеся при ПЦР небольшие фрагменты сложно детектировать после электрофореза в течение ночи при небольшом напряжении вследствие их интенсивной диффузии. Разрешение можно повысить, используя полиакриламидные или агарозные гели NuSieve (FMC Bio-Products, Rockland, USA) с высокой концентрацией агарозы (3—4%). Впрочем, если анализ нужно провести быстро и при небольших затратах, вполне приемлемы 2% агарозные гели.
Обычно при амплификации ДНК, выделенной из фиксированных тканей, выход ПЦР-продуктов ниже и они менее специфичны, чем в случае амплификации высокоочищенной ДНК.
Инструкция 1 по приготовлению агарозного геля (50мл).
Агарозный гель состоит из 1,8% агарозы и 98,2% Трис-буфера.
1. Взвешиваем агарозу (0,9г).
2. Переносим агарозу в термостойкую колбу.
3. Добавляем 50мл трис-буфера в колбу.
4. Устанавливаем плашку, устанавливаем гребенку на плашку.
5. Плавим агорозу путем нагревания в микроволновой печи до получения однородного раствора.
6. Добавляем краситель етидиум бромид в колбу и перемешиваем.
7. Заливаем агарозный гель на плашку.
4.3. Гибридизация ПЦР-амплифицированной ДНК по Саузерну
Этот метод позволяет идентифицировать полосы в геле, наблюдаемые после электрофореза амплифицированной ДНК. Для гибридизации используются как изотопно, так и неизотопно меченные зонды. Подробно метод гибридизации ПЦР-продуктов описан в протоколе 8.
Протокол 8. Гибридизация ПЦР-амплифицированный ДНК по Саузерну
Материалы
• Буфер для денатурации: 1,5 М NaOH, 0,5 М NaCl
• Буфер для нейтрализации: 1,5 М трис-НС1, 0,5 М NaCl, pH 6,5
• 20 х SSPE: 3,6 М NaCl, 0,2 М фосфат натрия, 0,02 М ЭДТА, рН 7,4
• 10% (в/о) ДСН
Методика
1.Для денатурации фрагментов ДНК гель вымачивают в буфере для денатурации при осторожном покачивании в течение 30 мин при комнатной температуре, затем выдерживают в буфере для нейтрализации в течение 30 мин.
2. За это время вымачивают найлоновый фильтр для переноса ДНК в 6 х SSPE в течение 5 мин (или дольше). Мы используем фильтры Genetrans 45 (Fiasco, Wolburn, Massachusetts, USA).
3. Собирают систему для переноса ДНК, и проводят перенос при комнатной температуре в течение ночи. В качестве буфера для переноса используют 20 х SSPE.
4. По окончании переноса снимают фильтровальную бумагу и отмечают положение лунок на фильтре. Фильтр снимают и для удаления прилипшей агарозы промывают в 20 х SSPE в течение 1 мин.
5. Пришивают ДНК к фильтру, поместив последний (стороной с ДНК вверх) на 5 мин под УФ-лампу (254 нм). Можно воспользоваться и специальным прибором для пришивания ДНК (Stratagene, La Jolla, California, USA).
6. Помещают фильтр в полиэтиленовый пакет и запаивают его. Проводят предгибридизацию при 42 0С в течение 10 мин и гибридизацию с соответствующим меченым зондом (гл. 13) при той же температуре в течение 30-60 мин. В качестве зонда обычно используют 5-меченные с помощью полинукле-отидкиназы олигонуклеотиды.
7. Промывают фильтр в двух-трех сменах 2 х SSPE, 0,1 % ДСН, по 5 мин в каждой смене. Иногда на этом этапе проводят отмывание и в более жестких условиях, но необходимость этой процедуры определяют опытным путем.
8. Гибридизовавшийся зонд выявляют с помощью радиоавтографии или неизотопными методами детекции.
4.4. Дот-блот-гибридизация ПЦР-амплифицированной ДНК
Дот-блот-гибридизация дает простой ответ по типу да-нет и особенно полезна в тех случаях, когда проводится анализ большого числа образцов. Один из примеров применения этого метода описан в протоколе 9.
Протокол 9. Дот-блот-гибридизация ПЦР-амплифицированнойДНК
Материалы
• Раствор для денатурации: 0,4 М NaOH, 1 мМ ЭДТА
• 20 х SSPE (см. протокол 15.7)
• Найлоновый фильтр
• Прибор для получения дот-блотов
Методика
1. Вымачивают найлоновый фильтр в дистиллированной воде не менее 5 мин.
2.10-20 мкл ПЦР-амплифицированной ДНК добавляют к 300 мкл раствора для денатурации (денатурация при этом происходит практически мгновенно).
3. Каждый образец вносят в отдельную ячейку прибора для получения дот-блотов; следят за тем, чтобы для всасывания образца из ячейки требовалось около 1 мин.
4. Промывают каждую ячейку 20 х SSPE три раза по 5 мин.
5. Вынимают фильтр из прибора и ополаскивают его в 20 х SSPE.
6. Выполняют операции, описанные в протоколе 15.7, начиная с п. 5.
5. Интерпретация результатов
Результат ПЦР можно квалифицировать как положительный или отрицательный в зависимости от того, обнаружена в образце интересующая нас последовательность-мишень или нет. Однако нарушение нормального хода амплификации, недостаточная чувствительность метода и непредвиденный полиморфизм последовательности-мишени в области связывания праймеров или гибридизационного зонда может дать ложноотрицательный результат. В случае загрязнения образцов и случайной гомологии между зондом, прай-мерами и последовательностью, сходной с мишенью, получаются ложноположительные результаты. Проблемы, которые возникают в связи с перекрестными реакциями с участием зонда или праймеров и возможным полиморфизмом.
5.1. Выбор контролей
Очень важным для правильной интерпретации результатов является выбор контролей. Положительные и отрицательные конт-роли должны быть хорошо охарактеризованы. Часто используют ДНК из клеточных линий, заведомо содержащих или не содержащих последовательность-мишень. В каждом анализе нужны как минимум три контроля:
• положительный контроль;
• отрицательный контроль;
• бланк-контроль.
Бланк-контроль — это реакционная смесь, в которой присутствуют все компоненты, за исключением ДНК; он является индикатором загрязнений. Один тип положительного контроля должен содержать максимальное число последовательностей-мишеней, другой — небольшое их число. Это позволяет определить чувствительность и эффективность ПЦР.
В некоторых случаях амплификация вообще не идет, и если есть основания думать, что полученный результат ложноотрицательный, очень трудно определить, по какой именно причине. Чтобы решить эту проблему, каждый образец необходимо тестировать, проведя амплификацию какой-нибудь геномной последовательности, например гена рецептора липопротеинов низкой плотности или р-глобинового гена. При этом размер геномной последовательнос!и-мишени должен быть немного больше, чем исследуемой; это позволит убедиться, что ДНК образца не слишком сильно деградирована. При ПЦР любого образца тканей человека геномная ДНК должна амп-лифицироваться. Отсутствие амплификации может означать инги-бирование фермента, сильную деградацию ДНК или то, что ее слишком мало. В этом случае надо повторить амплификацию образца, увеличив и уменьшив количество ДНК-матрицы или увеличив концентрацию ДНК-полимеразы Tag. Амплификация может отсутствовать несмотря на все предпринятые усилия. Это может быть связано с сильным некрозом исследуемой ткани или фиксацией препарата не в 10% формалине, а в других фиксаторах. В этом случае единственным выходом является повторная биопсия.
Геномные праймеры можно использовать в одном раунде ПЦР вместе с праймерами, специфичными в отношении последователь-: ности-мишени: амплификация двух последовательностей идет при этом одновременно (см. Протокол 4). Такой множественный ПЦР-тест может быть менее чувствительным, чем тот, в котором используется один набор праймеров, но он исключает появление ложноотрицательного результата.
5.2. Чувствительность
Определить чувствительность ПЦР-метода применительно к анализу фиксированных и залитых в парафин препаратов достаточно сложно. Это связано с отсутствием хорошо охарактеризованных катанных тканей с известным содержанием последовательности-мишени, а чувствительность метода при анализе очищенной ДНК и ДНК, полученной из фиксированных формалином тканей, при одних и тех же условиях заметно различается.
Однако в настоящее время есть возможность создавать искусственные фиксированные ткани с известным содержанием последовательности-мишени на основе хорошо охарактеризованных клеточных линий. Для этого смешивают клетки, содержащие нужную последовательность и не содержащие ее, смесь фиксируют в формалине и заливают в парафин. Полученный блок нарезают и обрабатывают так же, как биоптаты тканей (см. протоколы 1-4), проводят ПЦР и определяют чувствительность метода (рис. 8). И хотя этот прием не позволяет определить чувствительность ПЦР-метода с такой высокой точностью, как этого хотелось бы (см. ниже), он указывает путь к правильной интерпретации результатов.
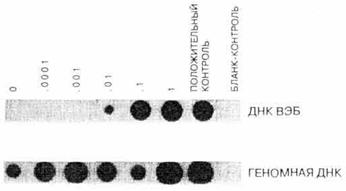
Рис. 8. Чувствительность ПЦР-анализа, проведенного на фиксированных формалином и залитых в парафин тканей. Смесь клеток Raji (примерно 50 копий ДНК EBV на клетку) и Molt 3 (не содержат EBV) собирали центрифугированием, фиксировали в 10% забуференном формалине в течение 1 ч и заливали в парафиновые блоки. Делали срезы толщиной 10 мкм, содержащие примерно по 1000 клеток, обрабатывали их, как описано в протоколах 1 и 2, и проводили ПЦР-анализ с использованием геномных и EBV-специфичных праймеров. Продукты амплификации анализировали с помощью дот-блот-гибридизации с геномным и EBV-специфичным зондами. Как и следовало ожидать, гибридизация с геномным зондом происходила во всех клеточных смесях, а с EBV-специфич-ным наблюдалась только в смеси, в которой доля клеток линии Raji составляла 0,01, но не 0,001 или 0,0001. Таким образом, при тех условиях, в которых проводилась ПЦР, удается выявить примерно 10 EBV-инфи-цированных клеток на 1000 нормальных (число клеток, с которым проводился ПЦР-анализ). Повысить чувствительность можно, изменив условия проведения ПЦР или используя для аначиза большее число клеток.
5.3. Количественный ПЦР-анализ
Логично думать, что количество образующейся при ПЦР ДНК коррелирует с числом последовательностей-мишеней в анализируемом образце. Однако так бывает не всегда. Часто такие исключения наблюдаются при работе с препаратами тканей, залитыми в парафин: в этом случае число факторов, влияющих на эффективность амплификации, особенно велико. Это могут быть природа ткани, способ фиксации и ее продолжительность, время, прошедшее от момента получения ткани до ее фиксации, «возраст» парафиновых блоков, размер среза, количество выделенной и использованной для ПЦР ДНК.
Часто клинические образцы получают в условиях, далеких от оптимальных, что тоже затрудняет проведение количественного анализа, особенно определение абсолютного количества последовательности-мишени. Для таких препаратов разработан простой относительный метод, основанный на ПЦР-анализе нескольких разведении препарата ДНК. Для этого готовят последовательные десятикратные разведения ДНК в воде и проводят амплификацию в условиях, позволяющих получить максимальную чувствительность; число циклов ПЦР при этом может достигать 50. Такой анализ позволяет оценить минимальное число последовательностей-мишеней в препарате, поскольку при достаточно сильном разведении амплификация должна прекратиться. Если параллельно проводить амплификацию какой-либо геномной последовательности, то можно определить соотношение между числом клеток и числом последовательностей-мишеней. При известном числе клеток в препарате (например, в препарате СМЖ) интерпретацию результатов проводят так, как это описано в табл. 7.
Таблица 7. Определение чувствительности методом разведении
| Число амплифицированных клеток |
Последователь-ность-мишень |
Геномная последовательность |
Интерпретация |
| 100000 |
+ |
+ |
1 мишень/100000 клеток |
| 10000 |
+ |
+ |
1 мишень/ 10000 клеток |
| 1000 |
+ |
+ |
1 мишень/ 1000 клеток |
| 100 |
+ |
+ |
1 мишень/ 100 клеток |
| 10 |
+ |
+ |
1 мншень/10 клеток |
| 1 |
+ |
+ |
Мишень практически в каждой клетке |
| 0 |
- |
- |
Система работает нормально |
Точное количество клеток в последних разведениях определить невозможно вследствие ошибок в пипетировании и пуассоновского распределения числа клеток. Геномные поcледовательности должны амплифицироваться и детектироваться на уровне примерно 1 клетки. В этом случае, если последовательность-мишень содержится в 1% клеток, последнее дающее положительный сигнал разведение должно соответствовать 100 клеткам.
6. Загрязнения
Способы предотвращения загрязнений препаратов, используемые в лабораторной практике, рассмотрены в работах [24, 25]. Очень важно хранить реактивы не целиком, а небольшими порциями, поскольку при обнаружении загрязнений приходится уничтожать все реактивы. Загрязнение может произойти в результате случайного внесения в образец посторонней последовательности-мишени. Это может случиться на любом этапе: при получении образца от больного, выделении ДНК, на стадии амплификации. Поскольку при ПЦР происходит экспоненциальное увеличение числа копий последовательности-мишени, то ложноположитель-ный результат может получиться при минимальном количестве посторонней ДНК, вплоть до одной молекулы. Поэтому при работе необходимо соблюдать такие же строгие меры предосторожности, как и те, что используются в микробиологии.
Если загрязнение все же произошло, то необходимо устранить его источник. Если загрязнен какой-то используемый в ПЦР реактив, то лучше выбросить все реактивы, поскольку попытки найти конкретный источник загрязнения приводят к большим затратам времени и грозят распространением посторонней ДНК. Если и после смены реактивов загрязнение сохраняется, то необходимо заменить или тщательно вымыть и обработать ультрафиолетом все оборудование (эксикаторы, микропипетки, микротомы). Для выявления загрязненного оборудования или загрязненных мест в лаборатории можно исследовать смывы, сделанные в соответствующих частях помещения [29]. Необходимо также тщательно проанализировать все этапы работы.
Если все эти меры не дают результата, есть два пути. Во-первых, можно сменить последовательность-мишень, либо подобрав праймеры к другой последовательности, либо использовав праймеры, которые гибридизуются с той же последовательностью-мишенью, но с внешней стороны от первой пары праймеров (обратная «гнездовая» ПЦР). Это предотвращает загрязнение ПЦР-амплифицированной ДНК. Во-вторых, можно полностью прекратить работу на несколько месяцев. Как это ни парадоксально, активные усилия по выявлению и устранению загрязнений ведут к их распространению (так называемое «ПЦР-сумасшествие» [30]), и только прекращение работ на 2-3 месяца приводит к самопроизвольному исчезновению загрязнения (по-видимому, в результате естественной деградации нуклеиновых кислот). При этом, конечно, можно продолжать работать с другими, не загрязненными последовательностями-мишенями.
7. Применение ПЦР в клинической лаборатории
Метод ПЦР может применяться для выявления в клинических образцах вирусов, микобактерий, простейших и бактерий, а также для обнаружения врожденных и приобретенных генетических нарушений и идентификации личности. Вопрос о возможной сфере применения ПЦР подробно рассмотрен в обзоре [31].
Широкое использование ПЦР в клинике сдерживается тем, что хотя сама процедура ПЦР полностью автоматизирована, подготовка и последующий анализ ПЦР-продуктов проводятся вручную и весьма трудоемки. Однако эти этапы тоже можно автоматизировать, и вероятно, вскоре будут созданы специальные приборы для проведения ПЦР-анализа, аналогичные современным биохимическим анализаторам, используемым в клинических лабораториях.
Промежуточным этапом в автоматизации ПЦР может служить использование наборов для ПЦР типа Amplitype (Cetus), предназначенных для типирования аллельных вариантов гена HLA DQ альфа. Располагая такими наборами, в предварительно подготовленные пробирки для ПЦР надо добавить только анализируемую ДНК и MgCl2. Праймеры в этом наборе биотинилированы, и амплифицированную ДНК легко детектировать после гибридизации с иммобилизованными зондами [8].
В настоящее время ПЦР-тесты можно ставить только в специально оснащенных клинических лабораториях. Образцы тканей, получаемые при обычных хирургических операциях, фиксируют формалином и заливают в парафин. Такие препараты можно исследовать методом ПЦР, что при необходимости позволяет провести молекулярно-генетический анализ без замораживания и хранения препаратов. После проведения обычных гистологических исследований, в большинстве случаев достаточных для постановки диагноза, некоторые препараты могут быть отправлены на ПЦР-анализ, позволяющий ответить на более специальные вопросы. Сфера применения ПЦР для целей диагностики будет постепенно расширяться, поскольку в клинической практике все чаще имеют дело с очень небольшими биоптатами (их получают, например, при эндоскопических исследованиях или это могут быть тонкоигольные аспираты и т. д.), обычный гистологический анализ которых затруднен. Перечислим вопросы, на которые можно получить ответ с помощью ПЦР.
а. Являются наблюдаемые нарушения привнесенными или в ткани действительно произошли неопластические изменения?
Это один из самых сложных вопросов. Часто проблема возникает вследствие слишком малых размеров препаратов или потому, что с ними обращались не должным образом (например, сдавливали, пересушивали и т. д.). Такие препараты можно без труда исследовать методом ПЦР и выявить в них те или иные молеку-лярно-генетические нарушения (некоторые из них перечислены в табл. 8). К сожалению, опыт применения методов ПЦР для диагностики заболеваний очень невелик, и в ряде случаев выявленные генетические нарушения оказывались характерны не только для опухолевой ткани, но и для нормальных тканей. Кроме того, генетические нарушения редко бывают строго специфичны для опухолей определенного типа. Тем не менее получаемые с помощью ПЦР данные могут использоваться для пополнения сведений, получаемых обычными гистологическими и другими методами, и позволяют более точно поставить диагноз.
Таблица 8. Некоторые генетические нарушения, выявляемые при помощи ПЦР
| Генетические нарушения |
Заболевания |
| Точковые мутации в гене K-ras |
Аденокарцинома • легких (у курящих) • поджелудочной железы • толстого кишечника |
| Точковые мутации в гене N-ras |
Острый миелолейкоз Миелодиспластический синдром Некоторые формы острого лимфолейкоза Миелома Фолликулярная лимфома Хронический миелолейкоз Лимфома Беркитта |
| Вирусные инфекции Вирус гепатита В Вирус папилломы человека |
Гепатоцеллюлярная карцинома Кондиломы, дисплазия и карцинома шейки матки Орофарингеальная неоплазия |
| HTLV-1 Вирус Эпштейна—Барр |
Т-клеточный лейкоз / лимфома у взрослых Лифома Беркитта, назофарингеальная карцинома, лимфома Ходжкина, неходжкинская лимфома |
б. Где находится первичная опухоль?
Нередко при биопсии берут кусочек ткани из области метастазов. При этом опухоль может быть диагностирована, но определить гистологическими методами ее первичную локализацию не удается. В то же время ПЦР-анализ метастазов позволяет выявить молекулярно-генетические нарушения, характерные для конкретных первичных опухолей. Так, если в лимфоузле обнаруживается вирус папилломы человека, то это свидетельствует об аногениталь-ной или орофарингеальной локализации первичной опухоли, а если вирус Эпштейна-Барр, то о назофарингеальной локализации.
в. Насколько полно излечено заболевание?
Морфологическая идентификация отдельных опухолевых клеток представляет большие трудности. Например, в биоптатах костного мозга редко удается выявить лейкемические клетки, если одна такая клетка приходится на 20 нормальных, а ПЦР-анализ легко позволяет сделать это. Небольшое число опухолевых клеток может присутствовать и в других органах и тканях — таких, как лимфатические узлы, периферическая кровь, СМЖ, моча, слизь. ПЦР используют для выявления последовательностей-мишеней, ранее обнаруженных в первичной опухоли (см. табл. 5), в других органах и тканях. Наличие таких последовательностей говорит о присутствии в организме опухолевых клеток и, следовательно, о неполном излечении. Однозначно установить наличие в организме таких клеток можно с помощью амплификации последовательностей-мишеней, строго специфичных для данной опухоли. Например, анализ иммуноглобулиновых генов с перестройками [32], рецепторов Т-клеток [33], хромосомных транслокаций [34, 35] позволяет выявить клональную природу клеток и тканей с неоплазией. Каково клиническое значение обнаружения очень Небольшого числа клеток, несущих специфичный для опухоли маркер, пока не вполне ясно. Можно лишь отметить, что ПЦР позволяет выявить минимальное число опухолевых клеток в организме и подобрать оптимальные методы лечения, когда какие-либо доклинические проявления заболевания еще отсутствуют.
8.Организация технологического процесса постановки ПЦР
Потенциально высокая чувствительность полимеразной цепной реакции делает совершенно необходимым особенно тщательное устройство ПЦР-лаборатории. Это связано с наиболее острой проблемой метода – контаминацией.
Контаминация – попадание из внешней среды в реакционную смесь специфических молекул ДНК, способных служить мишенями в реакции амплификации и давать ложноположительные результаты.
Такими мишенями могут быть продукты реакции, попадающие во внешнюю среду на этапе электрофореза из пробирок, в которых успешно прошла амплификация, либо специфическая ДНК из образцов на этапе пробоподготовки.
Существует несколько способов борьбы с этим неприятным явлением. Одним из них является использование фермента N-урацил-гликозилазы (УГ). В основе этого метода лежит способность УГ расщеплять молекулы ДНК со встроенным урацилом. Реакцию амплификации проводят с использованием смеси дНТФ, в которой дТТФ заменен на урацил, и после термоциклирования все образующиеся в пробирке ампликоны будут содержать урацил. Если до амплификации в реакционную смесь добавить УГ, то попавшие в реакционную смесь ампликоны будут разрушены, тогда как нативная ДНК останется целой и будет в дальнейшем служить мишенью для амплификации.
Другим способом инактивации ампликонов служит фотохимическое воздействия на молекулы ДНК. Для этого используют псорален или изопсорален, которые активируются кратковременным облучением ультрафиолетовым светом. Модифицированные этими соединениями молекулы ДНК не могут участвовать в реакции амплификации.
Однако, как известно, ни одна биологическая или химическая реакция не идёт со 100% эффективностью и, соответственно, после инактивации продуктов амплификации из миллиардов копий амплифицированного фрагмента хотя бы несколько останутся целыми, что существенно снижает ценность такого подхода. Кроме того, всегда остается риск кросс-контаминации от образца к образцу в процессе пробоподготовки.
Таким образом, оба эти метода лишь в некоторой степени позволяют устранить источник контаминации и не гарантируют от ложноположительных результатов.
Есть третий способ борьбы с результатами контаминации, рассматриваемый нами скорее как казусный – значительное уменьшение количества циклов реакции (до 25-30 циклов). Но даже при таком подходе риск получения ложноположительных результатов велик, т.к. и в этом случае при отсутствии ингибиторов легко получить продукт амплификации из-за контаминации.
Таким образом, несмотря на пользу преамплификационных мероприятий, направленных на инактивацию молекул ДНК, служащих причиной возникновения ложноположительных результатов, наиболее радикальным средством является заранее продуманная организация лаборатории.
Для уверенности в отсутствии контаминации необходимо каждую серию экспериментов сопровождать отрицательными контролями. В качестве отрицательных контролей мы рекомендуем использовать воду из комнаты пробоподготовки (после каждого десятого клинического образца желательно обрабатывать вместо биологического образца пробирку с водой).
Все реактивы рекомендуется хранить разлитыми на отдельные порции (аликвоты).
Если все же, несмотря на принятые меры, обнаружены следы контаминации, то необходимо все поверхности помещения, оборудования, пипеток и пр. обработать 0,1 М HCl, а все используемые порции реактивов следует заменить на новые.[20]
9.Устройство ПЦР-лаборатории
ПЦР-лаборатория должна быть разделена на три зоны:
1. Зона подготовки реакционной смеси («чистая зона»);
2. Зона пробоподготовки;
3. Электрофоретическая комната.
Все три зоны должны быть изолированными комнатами, снабженными предбоксниками. Полезно иметь устройство фильтрации воздуха.
Если первую и вторую зоны, в крайнем случае, допускается объединить (при наличии специальных боксов), то комната для электрофореза должна размещаться как можно дальше от двух других зон (другой этаж, другое здание) и иметь не связанную с другими зонами систему вентиляции. Это является одним из наиболее категорических требований при организации ПЦР-лаборатории.
Кроме того, желательно предусмотреть отдельные помещения для переодевания и хранения верхней одежды, приема пищи, складское помещение для лабораторных материалов.
Все производственные комнаты должны быть снабжены коротковолновыми ультрафиолетовыми лампами.
Перемещение пробирок, штативов и пр. должно производиться только в одном направлении, при этом потоки не должны пересекаться.

Клинический материал, поступивший в лабораторию, должен быть как можно быстрее обработан в комнате пробоподготовки. В эту же комнату должны поступать пробирки с реакционной смесью из «чистой зоны» для внесения в них препаратов ДНК. После этого пробирки помещают в амплификатор и, по окончании термоциклирования, не открывая крышек, переносят в комнату для электрофореза.
Все операции (пробоподготовка, подготовка реакционной смеси, электрофорез) должны выполнять разные люди.
Подготовку реакционной смеси следует проводить в ПЦР-боксе, снабженном электрическими розетками, лампами дневного и ультрафиолетового света.
Для обработки клинических образцов должны быть установлены ламинарные шкафы, обеспечивающие вертикальный поток воздуха для безопасности персонала при работе с инфекционным материалом, а также предусматривающие возможность работы без ламинарного потока и длительную экспозицию облучения внутренних поверхностей ультрафиолетовым светом.
Инактивацию биологического материала проводят в автоклаве в течение 1 часа при 1,5 атм. Допускается использование настольных автоклавов.
Запрещается внесение пробирок с положительными контролями или клиническими образцами как до, так и после обработки, в комнату подготовки реакционной смеси («чистую зону»).
Все производственные комнаты должны быть снабжены необходимым оборудованием и расходными материалами, в том числе халатами, закрепленными за соответствующими помещениями.[37]
10. Критерии выбора ПЦР-набора
С ростом популярности ПЦР-метода неизбежно растет и предложение тест-наборов, в том числе и отечественного производства. Неподготовленному потребителю довольно трудно разобраться в качестве того или иного предлагаемого на рынке диагностического набора, независимо от наличия прилагаемых документов и коммерческой активности фирмы-производителя. В такой ситуации можно лишь порекомендовать самим испытать разные тест-системы и сравнить их качество.
Критериями такой проверки служат, в первую очередь, специфичность и чувствительность тест-наборов. К сожалению, далеко не все тест-наборы имеют заявляемые характеристики.
Чувствительность можно проверить, приготовив несколько десятикратных разведений выделенной ДНК (например, из коллекционного штамма микроорганизма или из клинического образца, в котором достоверно имеется искомый возбудитель, что подтверждено культуральными методами исследований) и сравнив результаты амплификации. Если при этом один набор, уступая другому по чувствительности, дает больше положительных результатов на клинических образцах, следует задуматься о его специфичности.
Вероятна ситуация, когда положительные результаты, полученные после амплификации с использованием разных тест-систем, частично или полностью не совпадают. Производитель, защищая свою продукцию, может утверждать, что даже при отсутствии клинических признаков, получаемые положительные результаты лишь свидетельствуют о высокой чувствительности тест-системы. При несовпадении результатов с другим, более чувствительным, набором советуем все же убедиться, что выявляется именно искомый микроорганизм, а не что-то иное.
Следует также помнить, что наличие только одних клинических признаков не может служить доказательством присутствия предполагаемого микроорганизма. На первых порах для подтверждения качества используемой тест-системы рекомендуем сравнивать получаемые результаты с культуральными методами исследований.
Возможна и обратная ситуация, когда выигравшая по чувствительности тест-система недостаточно специфична. В этом можно убедиться, имея панель близкородственных микроорганизмов.
В некоторых случаях при использовании тест-системы, имеющей высокую чувствительность, возможно недовыявление положительных образцов. Это может быть в результате выбора праймеров в регионах ДНК, где возможны делеции. Иногда критичной для реакции становится замена нуклеотидов в местах отжига праймеров.
11. Определения точечных мутаций
Для определения точечных мутаций, которые составляют подавляющее большинство мутаций гена ФАГ рестрикция продуктов амплификации является одной из основных молекулярно-генетических методик. Ендонуклеази рестрикции (рестриктази), узнавая определенную последовательность ДНК, разрезают двойную спираль внутри или возле места узнавания сайта рестрикции. Полученные после рестрикции фрагменты ДНК разделяют в агарозном или полиакриламидном гелях. В многих случаях маркером мутации является наличие сайта рестрикции.
Для ДНК-диагностики мутаций R408W и ІVS10nt546, которые рассматриваются в данной работе, амплификация in vitro и последующая рестрикция продуктов амплификации является основной молекулярно-генетической методикой. Проведенный анализ первичной структуры участка 12 екзону гену ФАГ, соответствующего мутации R408W и рестрекционних сайтов известных специфических эндонуклеаз, позволяет определить, что мутантный сайт отвечает последовательности, которую узнает рестриктаза Sty, а также ее изошизомери Есоl301; BssTII; ЕсоТ141. Итак, наличие сайту рестрикции свидетельствует о присутствии мутации R408W [3]. Аналогично было установленное наличие сайта рестрикции для эндонуклеази Dde и ее изошизомеров при мутации IVS10nt546.
В случаях, если ген, ответственный за возникновение заболевания неизвестный, или прямые методы детекции мутаций оказались неэффективными для данной семьи (мутация не была идентифицирована), используют косвенные методы ДНК-диагностики. Косвенные методы направлены на анализ полиморфизмов в определенных фрагментах ДНК, но в отличие от прямых методов, они могут не входить в состав гена, необходимого для того, чтобы генетическое расстояние между изучаемыми фрагментами ДНК была небольшая. Понятно, что полиморфизм внутри этого фрагмента не определяет присутствие или отсутствие заболевания, тем не менее анализ таких полиморфных участков позволяет маркировать гомологические хромосомы и проследить их судьбу в поколениях отдельной семьи. Целью косвенных методов при диагностики рецесивних болезней есть решение вопроса – получил ли плод (ребенок) ту же пару хромосом от родителей, который и пробанд из этой семьи.
С точки зрения генетики любой фрагмент ДНК человека, последовательность нуклеотидов в которий может различаться в популяции, есть полиморфным локусом, а каждый вариант последовательности нуклеотидов этого фрагменту - одним из алелей данного локуса. Наиболее часто встречаются нуклеотидние замены - в среднем 1 на каждые 200-500 нуклеотидов. Такие полиморфизми есть биалельными. Более информативными есть так называемые VNTR и STR кластеры тандемних повторов, которые проявляют полиморфизм по количеству копий повторов. Определение алелели для такого вида полиморфизмов осуществляют с использованием метода ПЦР и последующим разделением продуктов амплификации в агарозному или полиакриламидному гелях. Результат разделения приведен на рис № 9.
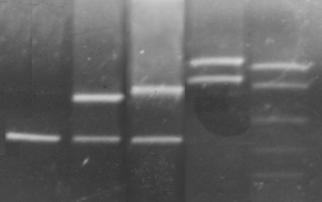
Рис. 9. Электрофореграмма продуктов амплификации минисателлитного VNTR –полиморфизма, 1.8% агарозний гель: 1 - генотип 3/3 (380/380 п.н.);2 - генотип 3/7 (380/500 п.н.); 3 - генотип 3/8 (380/530 п.н.); 4 - генотип 9/12 (560/650 п.н.); M - Маркер молекулярной массы ДНК плазмиды pBR322 гидролизированная эндонуклеазой MspI.
Для косвенного анализа мутации R408W при ДНК-диагностике фенилкетонурии используют анализ полиморфизма VNTR-локуса 3'-нетрансльованой области гену ФАГ. Для этого полиморфизма известно 7 алелей.
Использование выше приведенных методов позволяет проводить ДНК-диагностику фенилкетонурии и некоторых других заболеваний в семьях с высоким риском наследственных болезней. В особенности важное значение имеет возможность проведения пренатальной диагностики, даже в случаях, если мутации поврежденного гену в данной семье не идентифицированные, но семья информативная по полиморфной системе сцепленной с мутацией, или геном. Информативность определяется возможность маркировать с помощью полиморфизма мутантную и нормальную хромосомы родителей пробанда. То есть, если родители гетерозиготные по полиморфному маркеру, а пробанд гомозиготный по одному з алелей, то такую семью можно считать информативной по изучаемой системе. Если удается маркировать хромосомы одного из родителей, то система считается полуинформативной.
12. Denaturating Gradient Gel Electrophoresis
DGGE как диагностический инструмент.
Определение мутаций в DCR1,BCR2, MLH1, MSH2, MSH6, TP53, K-RAS,RET,RB, CFTR последовательностях.
ВСТУПЛЕНИЕ
DGGE основан на плавлении двух цепочечных фрагментов ДНК. Температура плавления (т.е. такая температура при которой любые две пары оснований в фрагменте теряют связи между собой) зависит от композиции пары оснований в фрагменте. Температура плавления различна даже в случае, когда одна пара оснований заменена. Путем добавления GC – богатых хвостов к праймеру который используется для амплификации достигается частичное расплетение двухцепочечной молекулы ДНК, при движении в геле с grad(C(денатурирующего агента)). GC хвост становится в обоих цепях. Такая структура позволяет значительно уменьшить электрофоретическую подвижность. Этот процесс проходит в различных местах геля при различных C(денатурирующего агента), когда одно или больше оснований заменено или удалено. Благодаря чему мутацию можно выявить. Используется grad(C(мочевины или формамида)). Определенная C(денатурирующего агента) в сочетании с температурой буфера равной 60 0С обладает эффектом как более высокая температура без присутствия, денатурирующего агента.

Рис. 10. Анализ мутаций в 12-м экзоне гена ФАГ методом DGGE. 12 % полиакриламидный гель, градиент денатуранта 20-60. 1 - R413P/норма; 2 – 5 - норма/норма; 6 - R408W/норма.
PCR следует за шагом температурной денатурации, при которой вся ДНК переходит в одно-цепочечное состояние. Когда гетерозиготная (т.е. такая у которой одна пара оснований замена) мутация присутствует отжиг позволяет, образовывается 4 различным фрагментам, два гомодуплексных и 2 гетеродуплексных. Предположим, он дает нормальную ДНК с G-C основаниями и мутантную с A-T парой оснований. Образуется два гомодуплексных молекулы ДНК, где G напротив C и A напротив T, а также 2 гетеродуплексных молекулы, где G напротив T и T напротив С. Эти 2 гетеродуплексные молекулы образуются только в случае гетерозиготных мутаций и останавливается выше в геле, потому что они слабее связаны энергетически.
Использование INGENYphorU электрофоретической системы в комбинации с DGGE фактически дает 100% уровень определения мутаций.
С целью постановки DGGE эксперимента были разработаны фрагменты ДНК. Для одного гена все фрагменты могут быть амплифицированы при помощи PCR. Использование геля длиной 20 см позволяет разделить до 5 различных фрагментов на одной дорожке.
Пример анализа мутаций в 12-м экзоне гена ФАГ методом DGGE приведен на рис. № 10.
13. Литература
1. Молекулярная клиническая диагностика. Методы. д.м.н. Г.И.Козинца к.м.д. П.А.Сломинского “МИР” 1999.
2. Нечипоренко М.В. Исследование мутаций и полиморфизмов последовательности ДНК гена фенилаланингидроксилазы в семьях высокого риска фенилкетонурии. Диссертация на соискание ученой степени кандидата наук. Киев 2002.
3. Нечипоренко М.В. Вивчення деяких мутацій гену фенилаланінгідроксилази в родинах з високим ризиком фенілкетонурії. Київ 1997. :
4. Innis M. A., Gelfand D. H., Sninsky J. J., While T. J. (ed.) (1990). PCR protocols, a guide to methods and applications. Academic Press, San Diego, California.
5. Erlich H. A. (ed.) (1989). PCR technology. Stockton Press, New York.
6. Promotional materials DGGE AS THE DIAGNOSTIC TOOL.
7. Ehlen T., Dubeau, L. (1989). Biochem. Biophys. Res. Commun., 160, 441.
8. SaikiR. K., Walsh P. S., Levenson C. H., Erlich H. A. (1989). Proc. Natl. Acad. Sci. USA, 86,6230.
9. ManosM. M., Ting Y., Wright D. K., Lewis A. J., Broker T. R., Wolinsky S. M. (1989). Molecular Diagnostics of Human Cancer, 7, 209-14. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
10. www.primer.ru/manuals/dna-tech/intro.htm
11. www.primer.ru/manuals/dna-tech/1.htm
12. GoeliS. E., Hamilton S. R., Volgelstein B. (1985). Biochem. Biophys. Res. Commun., 130,118.
13. Dubeau L., Chandler L. A., Gralow J. R., Nichols P. W., Jones P. A. (1986). Cancer Res., 46, 2964.
14. Greer C. E., Peterson S. L., Kiviat N. B., Manos, M. M. (1991). Am. J. Clin. Pathol., 95,117.
15. Shibata D., Martin W. J., Arnheim N. (1988). Cancer Res., 48, 4564.
16. www.primer.ru/manuals/dna-tech/2.htm
17. Wright D. K., Manos M. M. (1990). In PCR protocols: a guide to methods and applications (ed. M. A. Innis, D. H. Gelfand J. J. Sninsky, T. J. White), pp. 153-8. Academic Press, San Diego, California.
18. Shaunak S., Albright R. E., Klotman M. E., Henry S. C., Bartlett J. A., Hamilton J. D. (1990). J. Infect. Dis., 161, 1068.
19. Shibata D., Nichols P., SherrodA., RabinowilzA., Bernstein-Singer M., Hu E. (1990). Mod. Pathol., 3, 71.
20. www.primer.ru/manuals/dna-tech/5.htm
21. JacksonD.P.,BellS.,PayneJ.,LemsF.A.,SuttonJ., Taylor G. R., Quirke P. (1989). Nucleic Acids Res., 17, 10134.
22. www.primer.ru/manuals/dna-tech/3.htm
23. SambrookJ., Fritsch E. F., Maniatis T. (ed.) (1989). Molecular cloning. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY.
24. Kwok S. (1990). In: PCR protocols (ed. M. A. Innis, D. H. Gelfand, J. J. Sninsky, T. J. White), pp. 142-5. Academic Press, San Diego, California.
25. HiguchiR., Kwok S. (1989). Nature, 339, 237.
26. www.primer.ru/manuals/dna-tech/4.htm
27. Williams P., Simmond P., Yap P. L, Balfe P., Bishop J., Brettle R. et al. (1990). AIDS, 4, 393.
28. SarkarG., SommerS. S. (1990). Nature, 343, 27.
29. ConeR. W., HobsonA. C., HuangM. L. W., Fairfax M. R. (1990). Lancet, 336, 686.
30. See Walt Disney's Fantasia.
31. Eisenstein В. I. (1990). New Engl. J. Med., 322,178.
3.2. Yamada M., Hudson S., Tournay 0., Bittenbender S., Shane S.S., Long В., TsujimotoY., Caton A. J., Rovera G. (1989). Proc. Natl. Acad. Sci. USA, 86, 5123.
33. Loh E. Y., Elliot J. F., Cwirla S., LanierL. L., DavisM. M. (1989). Science, 243, 217.
34. Lee M., Chang K., Cabanillas F. et al. (1987). Science, 237, 175.
35. Cretena M., Seto M., Herzig G. P., Weiss P. D., Griffith R. C., Korsmeyer S. J. Proc. Natl. Acad. Sci. USA, 85,4869.
36. Shibala D., Namiki Т., Higuchi R. (1990). Am. J. Surg. Pathol., 14, 1076.
37. www.primer.ru/manuals/dna-tech/6.htm
