СОДЕРЖАНИЕ
стр. |
||
СПИСОК УСЛОВНЫХ СОКРАЩЕНИЙ |
4 |
|
ВСТУПЛЕНИЕ |
5 |
РАЗДЕЛ 1. ОБЗОР ЛИТЕРАТУРЫ
1.1. |
История исследования фенилкетонурии |
|
1.2. |
Картирование и клонирование гена фенилаланингидроксилазы |
|
1.3. |
Структура гена фенилаланингидроксилазы |
|
1.4. |
Белковый продукт гена ФАГ |
|
1.5. |
Полиморфные последовательности ДНК гена ФАГ |
|
1.6. |
Мутации гена фенилаланингидроксилазы |
|
1.7. |
Стадии ПЦР анализа |
|
| 1.8. | Выдиление ДНК матрици |
|
| 1.9. | Постановка ПЦР |
|
| 1.9.1. | Компоненты реакционной смеси |
|
| 1.9.1.1. | Праймеры |
|
| 1.9.1.2. | Фермент. |
|
| 1.9.1.3. |
dNTP |
|
| 1.9.1.4. |
Буфер |
|
| 1.9.1.5. |
Концентрация Mg2+. |
|
| 1.9.1.6. |
Анализируемый образец |
|
| 1.9.1.7. |
ПЦР добавка |
|
| 1.9.2. |
Температурный режим |
|
| 1.9.3. |
Число циклов |
|
| 1.9.4. |
«Эффект плато» |
|
| 1.9.5. |
Способ постановки ПЦР |
|
| 1.9.5.1. |
Способ постановки ПЦР с использованием «горячего старта» |
|
| 1.9.5.2. |
«Nested» ПЦР |
|
| 1.9.5.3. |
Полуколичественный анализ |
| 1.9.6. |
Контроль за прохождением реакции амплификации |
|
| 1.9.6.1. |
Положительные контроли |
|
| 1.9.6.2. |
Внутренние контроли |
|
| 1.10. |
Детекция |
|
| 1.10.1. |
Метод горизонтального электрофореза |
|
| 1.10.2. |
Метод вертикального электрофореза |
|
| 1.11. |
Проблема контаминации |
РАЗДЕЛ 2. МАТЕРИАЛЫ И МЕТОДЫ исследований |
||
|
2.1. |
Материалы исследования |
|
|
2.2. |
Методы исследований |
|
|
2.2.1. |
Выделение и очистка ДНК |
|
|
2.2.1.1. |
Выделение и очистка лейкоцитарной ДНК из образцов свежей крови |
|
|
2.2.2.2. |
Выделение ДНК из образцов крови длительного хранения, а также некультивированных клеток амниотической жидкости, хориона и плаценты. |
|
|
2.2.2. |
Амплификация некоторых последовательностей гена ФАГ методом полимеразной цепной реакции |
|
|
2.2.3. |
Идентификация мутаций методом рестрикционного анализа |
|
|
2.2.3.1. |
Амплификация in vitro последовательностей гена ФАГ для последующего рестрикционного анализа |
|
|
2.2.3.2. |
Рестрикционный анализ |
|
|
2.2.4. |
Анализ полиморфных последовательностей ДНК гена ФАГ |
|
|
2.2.4.1. |
Анализ аллельных вариантов VNTR-локуса 3`-нетранслируемого участка гена ФАГ |
|
|
2.2.4.2. |
Анализ аллельных вариантов STR-локуса (3-й интрон) гена ФАГ |
|
|
РАЗДЕЛ 3. РЕЗУЛЬТАТЫ ИССЛЕДОВАНИй |
||
|
3.1. |
ДНК-анализ мутаций R408W, IVS12nt1, R158Q, Y414C, IVS10nt546 гена ФАГ. |
|
|
3.1.2. |
ДНК-анализ мутаций в последовательности 7-го экзона гена ФАГ |
|
|
3.2. |
Оптимизпция условий проведения амплификации. |
|
|
РАЗДЕЛ 4. ВЫВОДЫ. |
СПИСОК УСЛОВНЫХ СОКРАЩЕНИЙ
|
БСА |
- |
бычий сывороточный альбумин |
|
ДНК |
- |
дезоксирибонуклеиновая кислота |
|
ЭДТА |
- |
этилендиаминтетрааминуксусная кислота |
|
ПААГ |
- |
полиакриламидный гель |
|
ПДРФ |
- |
полиморфизм длины рестрикционных фрагментов |
|
ПЦР |
- |
полимеразная цепная реакция |
|
РНК |
- |
рибонуклеиновая кислота |
|
ФАГ |
- |
фенилаланингидроксилаза |
|
ФКУ |
- |
фенилкетонурия |
|
DGGE |
- |
денатурирующий градиентный гель-электрофорез |
|
DHPLC |
- |
высокоэффективная жидкостная хроматография |
|
dNTP, дНТФ |
- |
дезоксинуклеотидтрифосфат |
|
STR |
- |
короткие тандемные повторы (short tandeme repeat) |
|
VNTR |
- |
варьирующее число тандемных повторов (variable number tandem repeat). |
|
п.н. |
- |
пар нуклеотидов |
|
т. п. н. |
- |
тысяч пар нуклеотидов |
|
kD |
- |
килодальтон |
|
Tris |
трис-(оксиметил)-аминометан tris(hydroxymethyl)aminomethane C4H11O3N |
ВСТУПЛЕНИЕ
В связи с расшифровкой последовательности ДНК генома человека на передний план выходит вопрос выяснения функциональной роли генов. Наиболее перспективным подходом в изучении генов человека, в условиях, когда проведение экспериментов ограничено особенностями исследуемого объекта, является анализ генов обуславливающих наследственные заболевания. Уже к 2000 году на хромосомах человека картировано около 1000 генов, мутации которых приводят к различным наследственным заболеваниям [1]. Одним из первых в геноме человека был картирован и идентифицирован ген фенилаланингидроксилазы, мутации в котором обусловливают развитие заболевания с аутосомно-рецессивным типом наследования – фенилкетонурии [2]. Изучение молекулярно-генетической природы этого заболевания позволяет исследовать частоту, природу и механизмы распространения мутаций в геноме человека. Анализ ассоциации генотипа и клинических проявлений заболевания отражает фенотипический эффект генетических изменений, а также служит основой для актуальных и перспективных исследований функционирования и взаимодействия генов.
С практической точки зрения, исследования молекулярно-генетических основ наследственных болезней человека открывает новые возможности в диагностике, профилактике и лечении тяжелых патологий наследственной природы. Реальностью сегодняшнего дня является применение программ генетического тестирования и пренатальной ДНК-диагностики, многих заболеваний, а делом ближайшего будущего – внедрение в клиническую практику методов генотерапии.
Цель и задачи исследования. Цель работы – разработка технологий проведения ПЦР для амплификации in vitro последовательности ДНК седьмого экзона гена ФАГ.
Для достижения цели были поставлены основные задачи исследования:
1. Освоить базовые технологии ДНК анализа.
2. Создать банк образцов лейкоцитарной ДНК больных и их родственников из семей высокого риска фенилкетонурии из разных регионов Украины.
3. Разработать эффективные условия проведения амплификации последовательности ДНК седьмого экзона гена ФАГ.
Объектом исследования является ДНК человека полученная из лейкоцитов периферической крови, а предметом – методы анализа мутаций в седьмом экзоне гена ФАГ.
В работе использовались методы: выделения и очистки ДНК, рестрикционный анализ, полимеразная цепная реакция, гель-электрофорез.
Научная новизна полученных результатов.
Была разработана математическая модель отжига праймеров которая позволила выбрать оптимальный температурный режим амплификации.
Практическое значение полученных результатов. Была разработана и оптимизирована методика для диагностики мутаций, локализованных в последовательности седьмого экзона гена ФКУ.
Разработана стратегия использования прямого анализа мутаций гена ФАГ, а также анализа сцепления мутантного гена с высокоинформативными ДНК-полиморфными маркерами для пренатальной диагностики, массового и селективного скрининга гетерозиготных носителей мутантных генов с целью профилактики этого тяжелого наследственного заболевания в Украине.
Автор высказывает искреннюю благодарность сотрудникам отдела генетики человека ИМБИГ НАНУ за постоянную поддержку и помощь во время выполнения работы.
Объем и структура работы. Диплом состоит из вступления, обзора литературы, описания материалов и методов исследований, полученных результатов собственных исследований и их обсуждения, заключения, выводов и перечня использованных источников. Текст диплома проиллюстрирован 60 таблицами и 28 рисунками. Дипломная работа изложена на 120 страницах машинописного текста.
Раздел 1
1.1. История исследования природы фенилкетонурии
Впервые в медицинской литературе описание этого заболевания появилось в 1934 году. [3]
Клинические симптомы фенилкетонурии появляются уже в первые месяцы жизни и могут быть разделены на такие группы: интеллектуальный дефект – умственная отсталость (чаще в степени имбицильности или глубокой дебильности); нейродегенеративные проявления; судорожный синдром [4].
Среди других симптомов выделяют: сходящееся и расходящееся косоглазие, асимметрия лица, снижение глоточного и нёбного рефлексов; вегетативно-трофические расстройства: сухость кожи, нарушение ее пигментации, ломкость волос, тенденция к артериальной гипотонии [5].
Основным диагностическим признаком заболевания является повышенная концентрация фенилаланина в крови, которая определялась ранее методом Гатри, а теперь методом иммунохимического анализа. В норме у детей этот показатель составляет 62±18 μM/л, а у больных ФКУ 120- 2400 μM/л [1]. В большинстве стран мира разработаны программы скрининга новорожденных на фенилкетонурию. В Украине эта программа действует, начиная с 1971. Благодаря этим программам не только стало возможным начинать пресимптоматическое лечение больных, но и определить частоту этого заболевания в разных странах. Так максимальная частота зарегистрирована в Ирландии 1:4500, а минимальная в Японии 1:120000 [6, 7]. В Украине на 8300 новорожденных рождается один ребенок больной ФКУ [8], причем в разных регионах частота ФКУ существенно отличается. Например, в Криворожской области - 1:12000, а в Полтавской и Черниговской областях - 1:4419. Частота носителей гена ФКУ в Украинской популяции 1:43 [9].
L-фенилаланин принадлежит к числу незаменимых аминокислот. В нормальных условиях лишь незначительная его часть используется для построения собственных белков, а большая часть превращается в тирозин [4].
В 1953 году Джервис и соавторы показали, что у больных фенилкетонурией преобразование фенилаланина в тирозин почти не происходит [10]. Позже было установлено, что причиной этого является отсутствие или значительное снижение активности лабильной (печеночной) фракции фермента фенилаланингидроксилазы [11]. Вследствие этого концентрация фенилаланина во всех жидких средах организма возрастает в десятки раз. В противоположность фенилаланину концентрация тирозина снижается [12].
Реакция превращения фенилаланина в тирозин осуществляется специальной ферментативной системой - гидроксилазы L-фенилаланина (ФАГ, фенилаланин 4-монооксигеназа, ЕС 1.14.16.1), которая катализирует присоединение гидроксильной группы к фенилаланину. В состав фермента входят два компонента: лабильный и стабильный [13, 14]. Стабильная фракция синтезируется во многих органах, в том числе почках, сердце, печени; лабильная фракция синтезируется только в печени [5]. В реакции преобразования фенилаланина в тирозин принимают участие кислород и тетрагидробиоптерин, который выступает в роли кофактора, а также дигидроптеридинредуктаза (ДГПР, КФ 1.6.99. 7) [13, 14, 15, 16, 17]. Реакция происходит по следующей схеме:
Эта реакция происходит при участии лабильной фракции фенилаланингидроксилазы. Стабильная часть ферментативной системы, а также ДГПР выполняют функцию восстановителя окисленного биоптерина в тетрагидробиоптерин [14].
У больных фенилкетонурией значительная часть фенилаланина выводится из организма с мочой, кроме того, для снижения концентрации фенилаланина происходит активация побочного пути преобразования аминокислоты – переаминирование. Около 10 % фенилаланина, поступающего с пищей, у больных фенилкетонурией выделяется с мочой в виде фенилаланина, 30 % - в виде фенилпировиноградной кислоты (ФПК), 20 % - фенилмолочной кислоты и 10 % - фенилуксусной [19].
Первичный дефект синтеза фенилаланингидроксилазы приводит к накоплению в организме человека фенилаланина и его производных. Следствием этого является ингибирование ряда ферментов, что влечет за собой вторичные нарушения обмена важных аминокислот, в первую очередь тирозина и триптофана [4].
Тирозин принимает участие в строительстве чрезвычайно важных белков организма, а именно меланина, норадреналина, адреналина, тироксина, дофамина [20, 21]. На первый взгляд видно, что дефицита тирозина, и как следствие веществ, которые из него получаются, при ФКУ не должно быть, поскольку тирозин поступает в организм с пищей. Однако фенилаланин и его аномальные метаболиты ингибируют ферменты, которые принимают участие в обмене тирозина и триптофана [21].
1.2. Картирование и клонирование гена фенилаланингидроксилазы
Работы по картированию гена ФАГ были начаты еще в 70-е годы. Первоначально, картирование проводилось на основе анализа сцепления с другими полиморфными белковыми маркерами человека. Было показано, что ген ФАГ сцеплен с геном PGM-1 (фосфоглюкомутаза), а также генами AMY-1 AMY-2 (амилазные локусы) картированными на хромосоме 1 [22, 23]. Однако, в 1979 году метод анализа сцепления был усовершенствован и повторно проведенные исследования сцепления с другими маркерами не подтвердили эту информации [24].
Точно локализовать ген ФАГ удалось лишь в 1984 году, когда стало возможно применять технологии рекомбинатных ДНК. Лидски и соавторы гибридизировали по Саузерну ДНК-панели клеточных гибридов человек/мышь с полученными к тому времени клонами кДНК гена ФАГ человека. Максимальный гибридизационный сигнал был получен для клонотеки хромосомы 12. В результате последующей гибридизации in situ с хромосомой 12, у которой были делетированы различные участки, ген ФАГ был точно картирован в области 12q22-24.1 [2].
В 1985 году Ву и соавторы, используя клонированную кДНК кролика в качестве специфической гибридизационной пробы, выделили и клонировали кДНК печени человека (клон phPAH47), на следующий год тем же коллективом авторов была определена полная последовательность нуклеотидов кДНК, а еще через год и полная последовательность гена ФАГ [25, 26].
Библиотека кДНК клеток печени человека, содержащая 14 миллионов рекомбинантов была сконструирована в клонируемом экспрессирующемся векторе lgt11. Для получения полной последовательности кДНК гена ФАГ авторами было проскринированo 100 000 рекомбинантов и получено 100 положительных гибридизационных сигналов. Четыре клона имели размер встроенного фрагмента 2000 - 2500 п.н., соответствующий полученному ранее методом гибридизации по Нозерну размеру мРНК фенилаланингидроксилазы. Эти 4 клона были обработаны рестриктазой EcoR1, встроенный фрагмент был извлечен и встроен по EcoR1 сайту плазмиды pBR322. Используя метод сиквенса, предложенный Максамом и Гилбертом, авторы определили размер и последовательность нуклетидов кДНК гена ФАГ человека [25, 26]
1.3. Структура гена фенилаланингидроксилазы
Ген фенилаланингидроксилазы имеет размер 90 т.п.н. В состав гена входит 13 экзонов и интронов [25]. Средняя длина экзонов приблизительно 114 п.н., самый короткий 9-ый экзон - 57 п.н., наиболее длинный 6-ой - 197 п.н. Тринадцатый экзон состоит из 894-х нуклеотидов, но протеинкодирующий участок содержит лишь 42 нуклеотида. Размеры интронов значительно больше: минимальная длина интрона 1т.п.н.(седьмой интрон), максимальная - 23.5 т.п.н. (третий интрон). В 3'-части гена сосредоточены более компактные интроны, в 5'-части - интроны больших размеров. Таким образом, суммарная длина экзонных последовательностей составляет 2.3 т.п.н., а интронных 85 т.п.н. Показатель соотношения некодирующей части гена к кодирующей для гена ФАГ один из наибольших в сравнении с другими известными на сегодня генами эукариот [25].
Было показано, что нуклеотидная последовательность гена фенилаланингидроксилазы человека имеют 90%-ю гомологию с аналогичным геном кролика [26] и 92,3% гомологию с геном ФАГ мыши [27], более того некоторые из описанных на сегодняшний день мутаций гена ФАГ человека идентифицированы и у мыши [28].
Экспрессия гена ФАГ происходит только в клетках печени. Незначительный уровень экспрессии отмечают уже в процессе эмбриогенеза, начиная с первого триместра беременности, но система гидроксилирования ФА включается в работу только в постнатальный период [29].
1.4. Белковый продукт гена ФАГ
Белковым продуктом гена ФАГ является железосодержащий фермент - фенилаланингидроксилаза, состоящий из 452-х аминокислотных остатков. Модель структуры фермента была создана в 1998-1999 г.г (рисунок 1.1) [30, 31]. Субъединицы фермента состоят из трех доменов: регуляторный домен (аминокислоты 1-142), каталитический домен (аминокислоты 143-406) и тетрамеризующий домен (аминокислоты 407-452) [29, 30, 31].
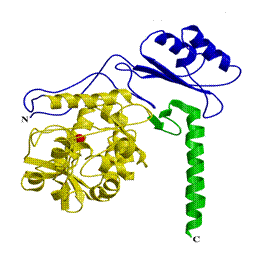
Рис. 1.1. Структура белка фенилаланингидроксилазы человека (модель). Красный - лиганд железа, желтый - католитический домен, фиолетовый - регуляторный домен, зелёный – тетромеризующий домен [30, 31].
1.5. Полиморфные последовательности ДНК гена ФАГ
При изучении молекулярной структуры гена ФАГ было идентифицировано несколько полиморфных участков ДНК, которые можно разделить на следующие группы: 1) биаллельные полиморфизмы длины рестрикционных фрагментов (ПДРФ); 2) мультиаллельные полиморфизмы, представленные в последовательности гена ФАГ минисателлитным VNTR (варьирующее число тандемных повторов) и микросателлитным STR (короткие тандемные повторы) полиморфизмами; 3) однонуклеотидные полиморфизмы (SNP).
Исторически первыми были идентифицированы ПДРФ полиморфизмы гена ФАГ. В процессе исследования кДНК было найдено восемь полиморфных сайтов рестрикции для семи разных эндонуклеаз: BglI, PvuII, EcoRI, XmnI, MspI, EcoRV, HindIII [32]. На рисунке 1.2 представлен весь спектр и локализация полиморфизмов гена ФАГ [33]. Точная локализация этих участков была проведена с помощью метода блот-гибридизации с кДНК-зондами, которые представляют собой разные участки гена.
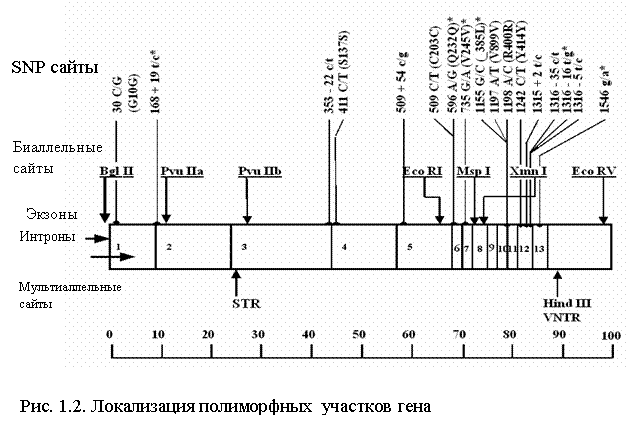
Полиморфизмы длины рестрикционных фрагментов (ПДРФ) ФАГ локуса образуют два кластера на 5'-конце и 3'-конце гена, расстояние между которыми приблизительно 30 т.п.н. В состав 5'-кластера входят экзоны 1-3 с полиморфными сайтами для эндонуклеаз BglI , PvuII (а), PvuII(б). Его размер составляет приблизительно 40 т.п.н. 3'-кластер с полиморфными сайтами для эндонуклеаз EcoRI , XmnI, MspI, EcoRV, HindIII включает экзоны 6-13 и имеет размер 30 т.п.н.[25]
При составлении рестрикционной карты гена ФАГ для HindIII полиморфизма, локализованного в 3`-части гена, было отмечено существование трех аллельных вариантов – явление не типичное для биаллельных ПДРФ полиморфизмов [32, 34]. Были определены такие размеры рестрикционных фрагментов, как HindIII 3,4 т.п.н.; HindIII 3,6 т.п.н. и HindIII 3,8 т.п.н. [35]. Для дальнейшего изучения этого участка гена ФАГ ДНК-зонды, содержащие 13-й экзон и 3 т.п.н. 3`-нетранслируемой последовательности проклонировали в ранее сконструированной плазмиде pKSP23E12. Это позволило более точно локализовать полиморфный регион и ограничить его участком размером 0,6 т.п.н [36]. При секвенировании этого фрагмента определили его первичную структуру и показали наличие АТ-богатого (70%) минисателлита на расстоянии 3 т.п.н от 13-го экзона в 3`-нетранслируемом участке. Размер повторяющейся единицы этого VNTR-полиморфизма составляет 30 п.н. Количество тандемных повторов варьирует от 3-х до 12-ти. Таким образом, полученные результаты объясняют наличие трех аллелей HindIII полиморфизма существованием минисателлитного VNTR полиморфизма в этом регионе, аллель с тремя повторами которого соответствует HindIII 3,4; 5; 6, 7, 8, 9 повторов – HindIII 3,6 и 12 повторов – HindIII 3,8 [36]. VNTR-полиморфизм обладает высокой информативностью [33, 37].
Для большинства мутаций гена ФАГ была показана ассоциация с определенными аллелями VNTR-полиморфизма, что свидетельствует о том что, какие бы события не привели к возникновению мультиаллельности в этом локусе они предшествовали процессу возникновения известных на сегодняшний день мутаций [36].
В транскрибируемой последовательности гена ФАГ идентифицировано 40 SNP полиморфизмов. Эти полиморфизмы представляют собой единичные нуклеотидные замены, но не являются ПДРФ. Девять SNP локализованы в интронных последовательностях и 31 - в экзонных [38]. Эти полиморфизмы называют молчащими, так как замена одного нуклеотида в аминокислоте при SNP полиморфизме не приводит к возникновению мутации, например Q232Q.
1.6. Мутации гена фенилаланингидроксилазы.
Как отмечалось выше, причиной развития ФКУ является дефект синтеза фермента ФАГ, а это, в свою очередь, является следствием мутаций в гене ФАГ. К 2001 году были идентифицированы более 600 мутаций, подавляющее большинство которых относится к классу миссенс мутаций, то есть мутации, которые приводят к единичной аминокислотной замене. Кроме миссенс мутаций есть нонсенс мутации, которые приводят к возникновению стоп кодона, а также делеции и мутации донорных и акцепторных сайтов сплайсинга [38].
К 2001 году было идентифицировано 140-ка мутаций гена ФАГ. Распростронение мутаций по месту локализации представлена в татлице 1.1 [38]. Частоты мутаций гена ФАГ у больных ФКУ в Украине представлена в татлице 1.2 [33, 39]. Так как наибольшее число мутаций находится в седьмом экзоне, то седьмой экзон требует особого внимания. Наш интерес к седьмому экзону усиливается тем фактом, что при помощи одной амплификации и 6 рестрикций мы можем индифицировать 6 мутаций (R261Q, G272X, P281L, R261X, S273F, R252W). Найбольший интерес представляет мажорная мутация в Украине и во многих странах Европы, R408W [33,40]. Также представляет интерес такие распространеные мутации как: Ivs12nt1, R158Q, Y414C, Ivs10nt546. Существует много методов анализа этих мутаций такие как: ПЦР, ИФА и блот гибридизация. Так как наименьшая себестоимость одного анализа, чувствительность и точность достигается при помощи полимеразной цепной реакции, то мы отдадим предпочтение именно ПЦР.
Таблица 1.1. Распространение мутаций по месту локализации.
|
Локализация |
Количество мутаций |
|
1 экзон |
7 |
|
3 экзон |
38 |
|
5-м экзоне |
1 |
|
7-м экзоне |
69 |
|
11-м экзон |
39 |
|
12 экзон |
22 |
|
10-м интрон |
1 |
|
12-м интрон |
1 |
Таблица 1.2. Частоты мутаций гена ФАГ у больных ФКУ в Украине
|
Мутация |
Частота % |
|
R408W |
57 |
|
R158Q |
3,7 |
|
R252W |
2,9 |
|
P281L |
2,3 |
|
Y414C |
1,6 |
|
Ivs10nt546 |
1,5 |
|
Ivs12nt1 |
1 |
|
R261Q |
1,2 |
|
G272X |
0,6 |
|
S237F |
0,6 |
|
R413P |
0,5 |
|
Не индефицированые |
27 |
1.7. Полимеразная цепная реакция
Полимеразная цепная реакция – это метод, имитирующий естественную репликацию ДНК и позволяющий обнаружить единственную специфическую молекулу ДНК в присутствии миллионов других молекул.
Открытие метода полимеразной цепной реакции (ПЦР) стало одним из наиболее выдающихся событий в области молекулярной биологии за последние десятилетия. Это позволило поднять медицинскую диагностику на качественно новый уровень.
Основные принципы использования праймеров (коротких искусственно синтезированных молекул ДНК) и состав ингредиентов, входящих в реакционную смесь для получения копий ДНК впервые были описаны Kleppe с соавт. в 1971 году. Однако тогда еще не была продемонстрирована основная черта ПЦР - экспоненциальное увеличение количества копий фрагмента исходной ДНК.
В 1983 году сотрудник фирмы «Cetus» Kary Mullis предложил метод, ставший в дальнейшем известным как полимеразная цепная реакция. Суть метода заключается в многократном копировании (амплификации) в пробирке определенных участков ДНК в процессе повторяющихся температурных циклов. На каждом цикле амплификации синтезированные ранее фрагменты вновь копируются ДНК-полимеразой. Благодаря этому происходит многократное увеличение количества специфических фрагментов ДНК, что значительно упрощает дальнейший анализ.
Следует отметить, что этому открытию сопутствовало развитие некоторых технологий. В частности, появление приборов, позволяющих автоматически синтезировать одноцепочечные фрагменты ДНК (олигонуклеотиды). В тот же период были обнаружены уникальные микроорганизмы, живущие в гейзерах. Их ферментативная система, в частности ДНК-полимераза, выдерживает высокие температуры горячих источников и сохраняет свою биологическую активность вплоть до 95°С, что является необходимым условием для проведения полимеразной цепной реакции [41].
Результатом открытия ПЦР стало почти немедленное практическое применение метода. В 1985 году Saiki с соавт. опубликовали статью, в которой была описана амплификация геномной последовательности ß-глобина [42]. С этого момента количество публикаций, в которых авторы сообщали о применении ПЦР в своих работах, стало увеличиваться в геометрической прогрессии. Метод приобрел такую популярность, что сегодня уже трудно представить работу в области молекулярной биологии без его использования. Особенно бурное развитие метод полимеразной цепной реакции получил благодаря международной программе «Геном человека». Были созданы современные лазерные технологии сиквенирования (расшифровки нуклеотидных последовательностей ДНК). Если в недавнем прошлом для расшифровки последовательности ДНК размером в 250 пар нуклеотидов (п.н.) требовалась неделя, то современные лазерные сиквенаторы позволяют определять до 5000 п.н. в день. Это в свою очередь способствует значительному росту информационных баз данных, содержащих последовательности ДНК различных биологических объектов. В настоящее время предложены различные модификации ПЦР, показана возможность создания тест-систем для обнаружения микроорганизмов, выявления точечных мутаций, описаны десятки различных применений метода.
Анализ выявления точечных мутаций на базе полимеразной цепной реакции состоит из следующих этапов:
- выдиление ДНК матрици;
- амплификация;
- детекция амплифицированых продуктов;
- полный или частичный анализ нуклеотидной последовательности продуктов ПЦР такими методами как: рестрикционный анализ, гетеродуплексный анализ, электрофоретическое фракционирование.
1.7.1. Компоненты амплификационной реакционной смеси
Для проведения полимеразной цепной реакции необходимо наличие в реакционной смеси ряда компонентов:
- Праймеры
- Taq-полимераза
- dNTP
- Буфер
- Mg2+
- Анализируемый образец
- ПЦР добавка
1.7.1.1. Праймеры – искусственно синтезированные олигонуклеотиды, имеющие, как правило, размер от 15 до 30 п.н., идентичные соответствующим участкам ДНК-мишени. Их можно синтезировать с помощью автоматических синтезаторов ДНК. Часто в 5'-концевой участок праймеров для упрощения клонирования ПЦР-амплифицированной ДНК вводят сайты узнавания для рестриктирующих эндо-нуклеаз. В 3'-концевой участок праймеров не могут быть внесены никакие замены, так как при низкой концентрации нуклеозидтрифосфатов такое нарушение блокирует амплификацию. Если концентрация нуклеозидтрифосфатов находится на уровне, обычном для ПЦР то небольшое нарушение гомологии между 3'-концом праймера и матрицей не сказывается на ходе реакции. Таким образом, с помощью ПЦР можно различить аллели, различающиеся всего одним нуклеотидом.
Подбор праймеров - ключевое звено ПЦР, поскольку именно ими определяется возможность амплификации и выявления нужной последовательности, а также чрезвычайная гибкость метода. Простое варьирование праймеров позволяет выявлять многие патогенные микроорганизмы и генетические нарушения при минимальных изменениях в методике [41].
Необходимо отметить, что праймеры могут отжигаться друг с другом, образуя праймер-димеры и могут сами на себя, образуя петли [43]. И то, и другое приводит к значительному расходу праймеров на синтез побочных (неспецифических) продуктов реакции и, как следствие, значительно уменьшает чувствительность системы. Это затрудняет или делает невозможным чтение результатов реакции при проведении электрофореза [44].
ПЦР-амплифицированные фрагменты ДНК имеют разный размер. Он определяется суммой размера праймеров и расстояния между их 3'-концами и в большинстве случаев лежит в диапазоне 100 - 300 нуклеотидов. Скорость репликации с помощью ДНК-полимеразы Tag составляет 35 - 100 нуклеотидов в секунду [47].
Оценка количества праймера может быть определина согласно следующего алгоритма. Если в 100µl будет синтезировано 3µg 500bp продукта, то на это должно пойти праймера: 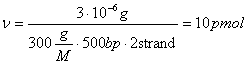 . Используемый диапазон концентраций: 0.1-0.6 µM.
. Используемый диапазон концентраций: 0.1-0.6 µM.
Лучше, если пара "сбалансирована", т.е. разница температур плавления не превышает 2-4oС.
При создании ПЦР-тест-системы одной из основных задач является правильный подбор праймеров, которые должны отвечать ряду критериев:
1. Праймеры должны быть специфичны. Особое внимание уделяют 3'-концам праймеров, т.к. именно с них Taq-полимераза начинает достраивать комплементарную цепь ДНК. Если их специфичность недостаточна, то, вероятен синтез неспецифической ДНК (коротких или длинных фрагментов). Они видны на электрофорезе в виде тяжелых или легких дополнительных полос, иногда шмеров, выглядящих сплошным мазком на электрофорезе. Это мешает оценке результатов реакции, т.к. легко перепутать специфический продукт амплификации с синтезированной посторонней ДНК. Часть праймеров и дНТФ расходуется на синтез неспецифической ДНК, что приводит к значительной потере чувствительности.
2. Праймеры не должны образовывать димеры и петли, т.е. не должно образовываться устойчивых двойных цепей в результате отжига праймеров самих на себя или друг с другом.
3. Область отжига праймеров должна находиться вне зон мутаций, делеций или инсерций в пределах видовой или иной, взятой в качестве критерия при выборе праймеров, специфичности. При попадании на такую зону отжига праймеров происходить не будет, и, как следствие, будет получен ложноотрицательный результат.
Праймеры играют ключевую роль в образовании продуктов реакции амплификации. Правильно подобранные праймеры обеспечивают специфичность и чувствительность тест-системы.
1.7.1.2. Фермент.
В 1980 году полимеразная активность была открыта у некоторых форм термофильных бактерий. Таg-полимераза была выделена из бактерий вида Thermus aquaticus, способных расти при температуре 70-75°С. Молекулярный вес очищенного протеина 94 кД и оптимальная температура полимеразной активности 70 - 80°С. При 90°С активность фермента уменьшается, но полимераза не денатурируется, при снижении температуры до 70-80°С уровень активности восстанавливается.
Tag-полимераза имеет очень высокую скорость синтеза. При оптимальных условиях фермент может достраивать до 150 основ на секунду. При низкой температуре активность падает до 2 основ на секунду. Время полужизни Таg-полимеразы при 95°С составляет 40 минут.[2]
Для проведения ПЦР-тестов обычно используют ДНК-полимеразу, выделенную из Thermus aquaticus. У этого фермента отсутствует корректирующая 3'-4'-5'-активность и частота ошибок при считывании достигает 1/10 000 нуклеотидов. Этой точности (хотя она и ниже, чем у многих других полимераз) достаточно для того, чтобы возникающие при репликации последовательности-мишени ошибки не создавали особых проблем.
Оптимальное количество фермента зависит от количества матрицы и длины ПЦР продукта.
1U – это то количество фермента которое связывает 10 нмоль дезоксинуклеотидтрифосфатов за 30 мин. при 74ºС [48].
Если используется слишком маленькое количество Taq, выход продукта снижается, причем это снижение тем серьезнее, чем больше размер продукта. Не стоит использовать и слишком большой избыток Taq полимеразы, так как при этом вначале появляются высоко- и низкомолекулярные шмеры (превышение ~2-4 раза), а затем весь продукт становится чрезвычайно маленьким (превышение ~4-16 раз).
Для рутинного использования часто используют смесь Taq:Pfu=100:1, у которой точность сиетеза практически такая же, как у Pfu полимеразы. В удобстве использования такая смесь не уступает Taq полимеразе.
Так как для Ampli Taq "Perkin-Elmer" Mw=94kDa и 250 000u - 1 mg то 1u соответствует 4ng, или 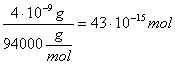 =43fmol.
=43fmol.
Taq pol. обладает 5'-3' экзонуклеазной активностью. Эта активность может вызвать проблемы, связанные с деградацией 5' концов продукта.
Точность Taq-pol зависит от концентрации Mg2+, dNTP's, сбалансированности dNTP's, pH. В среднем, частота ошибок чуть ниже, чем 1 замена на 100-300bp.
Так как Taq полимераза с высокой частотой включает неправильные нуклеотиды, то для правильного определения последовательности нужно сиквенировать не одну клонированную матрицу, а несколько. Это касается только клонированных PCR-продуктов. Если PCR-продукты сиквенировать непосредственно, то последовательность получается точной, так как для того, чтобы обнаружить мутацию, она должна присутствовать в >10% матриц. Это возможно лишь в том случае, если мутация произошла в первом цикле, а матриц при этом было не более 5 штук.
Для Phusion Polymerase оптимальное количество полимеразы, как правило, лежит в пределах 0,5 – 2 U на 50 мкл [48].
1.7.1.3. dNTP Смесь дезоксинуклеотидтрифосфатов (дНТФ) – дезоксиаденозинтрифосфата (дАТФ), дезоксигуанозинтрифосфата (дГТФ), дезоксицитозинтрифосфата (дЦТФ) и дезокситимидинтрифосфата (дТТФ) – «строительный материал», используемый Taq- полимеразой для синтеза второй цепи ДНК. Важно поддерживать концентрацию значительно выше, чем Km соответствующей полимеразы. Чем меньше концентрация dNTPs, тем выше точность синтеза. Диапазон используемых концентраций составляет 50-500 µM. Высокие концентрации дНТФ могут привести ко включению не комплементарных нуклеотидов в следствии термодинамического безразличия какой нуклеотид включить в удленяющуюся последовательность [49]. Смесь дезоксинуклеотидтрифосфатов обладают значительной буферной емкостью, которая уменьшается с каждым циклом амплификации [44]. Для Phusion™ DNA Polymerase рекомендуется использовать такое количество дезоксинуклеотидтрифосфатов, чтобы их концентрация в амплификационной смеси составляла 20 мкМ [48].
1.7.1.4. Буфер.
Буфер - смесь катионов и анионов в определенной концентрации, обеспечивающая оптимальные условия для реакции, а также стабильное значение рН [42]. Многие полимеразы имеют свои буфера. Буфера также различаются по предназначению. Например компания Finnztmes рекомендует использовать разные буфера в зависимости от соотношения AT к GC. Так как уровень ошибок полимеразы с GC буфером (9,5.10-7) гораздо выше, чем полимеразы с HF буфером (4,4.10-7), GC буфер рекомендуется использовать, когда HF не работает [48].
1.7.1.5. Концентрация Mg2+.
Так как ДНК полимераза магний - зависимый фермент, то концентрация ионов Mg2+ является важным параметром амплификации [48]. На молекулярном уровне: Mg2+ образует комплексы с dNTP's (именно эти комплексы являются субстратом для Taq pol). C Mg2+ стехиометрически связываются dNTP's, EDTA, (PO4)3-. Так как повышение концентрации Mg2+ вызывает повышение температуры плавления DNA [44, 50], то повышение концентрации Mg2+ приводит к стабилизации комплексов ДНК-праймер с неполной гомологией, что снижает специфичность амплификации[48]. Избыток ионов стабилизирует двухцепочечную ДНК и предотвращает полную денатурацию ДНК. Процесс денатурации ДНК рассмотрен в работе [51], а также в разделе 1.7.2. Оптимальная концентрация ионов Mg2+ зависит от концентрации дезоксинуклеотидтрифосфатов, ДНК-матрицы и состава буфера. В общем случае для стандартной ПЦР оптимальная концентрация ионов Mg2+ лежит в пределах от 0,5 до 1 мМ больше общей концентрации дезоксинуклеотидтрифосфатов. Если праймеры и (или) ДНК содержат хелаты, такие как EDTA или EGTA, концентрация Mg2+ должна быть изменена в сторону увеличения. В случае необходимости дальнейшей оптимизации условий, рекомендуется увеличивать концентрацию Mg2+ с шагом 0,2 мкМ. [48]. Слишком низкая концентрация Mg2+ обуславливает низкий выход продукта, в то время как слишком высокая концентрация Mg2+ приводит к снижению выхода ПЦР-продукта. На рисунке № 1.3. представлено влияние концентрации ионов Mg2+ на амплификацию. Для данной системы оптимальная концентрация ионов Mg2+ составляет 2,5 млмоль/л [50]. Также необходимо отметить, что высокая концентрация Mg2+ может привести к неспецифической амплификации [44].
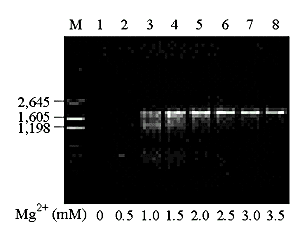
Рисунок № 1.3. Влияние концентрации ионов Mg2+ на амплификацию.
1.7.1.6. Матрица.
Матрица это образец, подготовленный к внесению в реакционную смесь, который может содержать искомую ДНК, служащую мишенью для последующего многократного копирования. При отсутствии ДНК-мишени специфический продукт амплификации не образуется.
В качестве матрицы может выступать:
1. Геномная DNA млекопитающих < 0.5-1µg;
2. Геномная DNA бактерий ~1-10ng;
3. DNA фага лямбда. Достаточно 1µl из 200µl элюции одиночной бляшки;
4. PCR продукт разбавленный в ~ 106раз.
5. Плазмидная DNA:
5.1. 0.1-1.0µl ночной культуры,
5.2. ~50ng очищенной суперскрученной DNA (такое большое количество берется из-за того, что pDNA - кольцевая и при понижении температуры она ренатурирует, не оставляя праймеру шанса связаться с ней. Если хочется уменьшить количество вносимой pDNA, нужно проводить предварительную щелочную денатурацию. Для этого необходимо взять 4µl pDNA и смешать с 1µl 1N NaOH, полученый раствор выдерживают при 65-85oC на протяжении 5 минут и резко охлаждают до 0oС (или заморозить) и добавить 1µl 1N соляной кислоты.
1.7.1.7. ПЦР добавка.
ПЦР добавки - вещества, улучшающие специфичность (и/или) выход PCR реакции. Для большинства амплификаций ПЦР добавки не используются. При амплификации GC-богатых участков, некачественных образцов ДНК и амплификации фрагментов больше 1 т. п. н. рекомендуется использовать вспомогательные вещества, которые приведены в таблице № 1.4[44].
Важно отметить, что добавление этих веществ к уже оптимизированной и хорошо идущей реакции может вызвать ухудшение выхода. Это объясняется тем, что - указанные вещества изменяют взаимодействие между праймерами и матрицей и между матрицей и полимеразой.
Таблица № 1.4.
|
Вещество: |
Сток: |
Используемые концентрации: |
Механизм действия: |
|
Acetamide |
25% |
~5% |
повышение растворимости |
|
Betain Na |
5M |
0.5-2M |
стабилизация фермента, понижение Tm DNA, выравнивание Tm AT- и GC-богатых последовательностей |
|
BSA |
10 µg/µl |
~0.1 µg/ml |
стабилизация фермента |
|
DMSO |
100% |
2.0-15% (оптимально ~3%) [48] |
Помогает денатурировать CG-богатую ДНК матрицу. 10% DMSO снижает температуру плавления на 5,5-6,0ºС [52] |
|
Formamid |
100% |
1-5% |
|
|
Glycerol |
100% |
5-20% |
стабилизация фермента |
|
NP-40, Tween 20, Triton X-100 |
10% |
0.1-0.5% (улучшает seq. PCR) |
стабилизация фермента |
|
PEG 8000 |
40% |
5-20% |
повышение эффективной концентрации |
|
Tetramethylammonium chloride (TMA chloride) |
5M |
0.01-0.1mM |
повышение специфичности |
|
Pyrophosphatase thermostable |
5 u/µl |
0.001-1u/реакцию |
устранение пирофосфатов, которые могут обращать реакцию полимеризации |
|
ssDNA binding protein E.coli |
500 µg/ml |
5 µg/ml |
стабилизация ssDNA |
|
ssDNA binding protein T4 gene 32 protein |
500 µg/ml |
улучшает выход в концентрациях 0.5-1.0mM. |
стабилизация ssDNA |
Таблица № 1.5.
|
Вещество |
Максимальная |
90% ингиби-рования |
|
|
эффективность |
специфичность |
||
|
TMA chloride |
1.9(5 mM) |
0.5(20 mM) |
(35 mM) |
|
TMA oxalate |
2.2(2 mM) |
1.0(2 mM) |
(9 mM) |
|
TMA acetate |
1.0(<10 mM) |
0.4(20 mM) |
(40 mM) |
|
TMA hydrogen sulfate |
1.2(0.5 mM) |
1.0(50 mM) |
(70 mM) |
|
Ammonium chloride |
1.0(<20 mM) |
0.2(<20 mM) |
(50 mM) |
|
Benzyldimethyl-hexadecylammonium chloride |
1.1(0.2 mM) |
0.4(1 mM) |
(1.0 mM) |
|
HTA bromide |
1.2(0.05 mM) |
0.5(0.5 mM) |
(0.8 mM) |
|
HTA oxalate |
1.0(<0.2 mM) |
0.28(<0.2 mM) |
(0.5 mM) |
|
Betaine monohydrate |
1.1(100 mM) |
0.4(750 mM) |
(900 mM) |
|
Dimethyl sulfoxide |
1.0(<1.4 M) |
0.6(1.4 M) |
(1.6 M) |
|
Formamide |
1.4(0.5 M) |
0.8(1 M) |
(2.0 M) |
|
PCR без добавок |
1.0 |
0.2 |
|
В скобках – концентрация веществ, приводящая к указанному эффекту. Число перед скобками означает относительную эффективность действия. Специфичность оценивается как доля DNA, приходящаяся на специфическую полосу. TMA производные практически не изменяют оптимальную температуру отжига [53].
Другие вещества. В таблице № 1.6. приведены вещества, которые могут попасть в амплификационную систему и их действие.
Таблица № 1.6.
|
Вещество |
Действие |
|
Агароза |
не мешает до ~1%. |
|
АА акриламид линейный |
не мешает (по крайней мере в низкой концентрации). |
|
AcONa (pH 5.0) |
начинает ингибировать при>5mM. |
|
BSA ацетилированный |
10µg/ml – ингибирует |
|
BSA heat инактивированный |
10µg/ml - не мешает |
|
Vanadil ribonucleoprotein complexes |
ингибируют при > 0.4mM; не мешают при <0.04mM (когда цвет 8-гидроксихинолина в феноле не изменяется при экстракции - уже не ингибируют) |
|
Гемоглобин (концентрация в крови ~160mg/ml) |
уменьшает выход при >1mg/ml |
|
Гепарин |
ингибирует (используется для предотвращения свертывания крови 14.3 u/ml, не устраняется органическими растворителями и переосаждением). |
|
EDTA |
связывается с Mg2+ стехиометрически, начинает ингибировать при >0.5mM, PCR не идёт при 1mM. |
|
DEPC |
ингибирует реакцию, лучше не использовать в PCR-реакции растворы, обработанные DEPC |
|
Желатин |
10µg/ml - не мешает |
|
Изопропанол |
ингибирование при концентрациях >1% (более сильный ингибитор, чем этанол) |
|
Масло, покрывающее PCR |
может устранять ингибирующий эффект некоторых загрязнений |
|
Мочевина (в моче ~350mM) |
на разные реакции - по разному, в среднем, начинает ингибировать при >20mM |
|
NaCl |
заметно ингибирует при 25mM, PCR не идёт при 50mM |
|
RT смесь |
не влияет на PCR реакцию при концентрации 10%, начинает ингибировать при >15% |
|
RNA |
ингибируют (>0.5µg на 20µl PCR); ингибирующий эффект можно устранить добавлением в реакцию RNase (термостабильна) |
|
Сахароза |
не мешает до 30%. |
|
Фенол |
уменьшает выход при >0.2%, PCR не идёт при 0.5% |
|
Ficoll 400 |
не мешает до 1% (выше - ингибирует) |
|
HxPO4 |
Mg3(PO4)2 - образует преципитат при NT, если >0.25mM (в PBS - HxPO4~9.3mM) |
|
Этанол |
для некоторых реакций - стимулирующий эффект при 1%, для других - ингибирование при концентрациях >1% |
|
Хлороформ |
не влияет на PCR реакцию даже при концентрации 5%. |
|
Цитрат Na |
не оказывает влияния при 1mM |
|
Меланин |
Заметно от 0.01µg/µl, ингибирует: 0.08µg/µl |
|
Polyvinilpyrollidon (PVP) |
не ингибирует по крайней мере до 2% |
1.7.2. Температурный режим.
В процессе реакции амплификации с ДНК происходит ряд событий, которые обеспечиваются определенными температурными циклами. Каждый цикл амплификации состоит из трех этапов: денатурация, отжиг праймеров, элонгация, которые будут рассмотрены далее.
Многие иследователи используют 2-5 минутный предварительный нагрев. Предполагается, что это позволяет денатурировать ДНК матрицу лудчше.
1. Предварительный нагрев.
Предварительный нагрев проводят для прогрева пробы и разрушения двухцепочечной ДНК. Температуру и время предварительного прогрева выбирают исходя из состава ДНК-матрицы и свойств полимеразы. Для Tag полимеразы рекомендуется проводить предварительный прогрев до 94ºС на протяжении 30 - 300с. Экспозиция 300с используется для амплификации CG-богатых участков. Однако, если доля CG-оснований в ПЦР продукте превышает 75%, то рекомендуется использовать 98ºС. Также как все молекулы ДНК не плавятся одновременно, так и каждая отдельно взятая молекула не плавится вся [54]. Молекула ДНК плавится постепенно рисунок № 1, и в первую очередь в участках с высокой долей AT оснований с образованием свободных концов и внутренних циклов, которые представлены на рисунке № 1.5. Вероятность образования внутреннего цикла (рис. 1.6.) рассмотрена в работе [55].
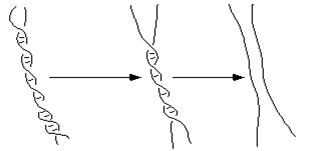
Рисунок № 1.4. Процесс денатурации двойной спирали.
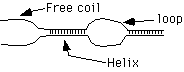
Рисунок № 1.5. Образование свободных концов, спирали и внутренних циклов при денатурация двойной спирали [55].
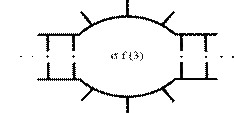
Рисунок № 1.6. Образование внутреннего цикла.
Однако использование высокой температуры имеет ряд недостатков. Один из основных недостатков - это снижение времени полураспада полимеразы. Время “полураспада” Taq полимеразы в зависимости от температуры представлены в таблице № 1.7. Зависимость времени “полураспада” Taq полимеразы от буфера при 96.5oC представлена в таблице № 1.8.
Так при 98ºC время полураспада Tag Pol составляет всего 5 мин, что обуславливает необходимость ее замены. В таких случаях рекомендуется использовать Gold DNA Polymerase или Phusion DNA Polymerase. При использовании Phusion DNA рекомендуется проводить 30-180 секундный предварительный прогрев при температуре 98ºС. Если проводить массовый скрининг CG богатой области, то использование Gold DNA Polymerase или Phusion DNA Polymerase может зтачительно повысить себестоимость исследования.
Таблица № 1.7. Время "полураспада" Taq полимеразы в зависимости от температуры.
|
Температура oС |
Время мин |
|
92.5 |
130 |
|
95.0 |
40 |
|
97.5 |
5-6 |
Таблице № 1.8. Скорость разрушения Taq полимеразы в различных буферах при 96.5oC.
|
Буфер |
Время "полураспада" мин |
|
H2O |
2.5 |
|
TE |
7.5 |
|
20mM Tris, pH9.0 |
10 |
Альтернативным вариантом может быть использование смеси Gold DNA Polymerase или Phusion DNA Polymerase с Tag-полимеразой при этом необходимо использовать денатурирующие агенты такие как бетаин, DMSO или другие. Использование таких веществ позволяет снизить температуру денатурации CG-богатого участка до 94ºС.
Если матрица одноцепочечная или PCR-продукт, то предварительный прогрев можно не проводить.
Так как кольцевая pDNA умеет ренатурировать после тепловой денатурации, то предварительный нагрев рекомендуется проводить при 95oC на протяжении 1 мин. Данные условия являются компромиссом при котором, с одной стороны, некоторая часть плазмид никуется и превращается в одноцепочечные продукты, с другой стороны, не слишком большая часть матриц повреждается.
2.1. Денатурация. На первом этапе необходимо расплести двойную цепь ДНК, находящуюся в образце.
Температура и время денатурации выбираются как компромисс между двумя целями:
- "хорошо" денатурировать матрицу,
- не сильно повредить матрицу и Taq полимеразу [56].
Время денатурации сильно зависит от типа используемых пробирок. Для тонкостенных 0.2 ml пробирок достаточно 10-15 секунд при температуре 94oС [44].
2.2. Отжиг праймеров.
На втором этапе праймеры присоединяются к одноцепочечной ДНК-мишени. Этот процесс носит название «отжиг» (от англ. «annealing»).
Оптимальная температура и время отжига праймеров обеспечивают эффективную ПЦР. Если температура отжига праймеров будет занижена, то это может привести к неспецефической амплификации, а если завышена, то - к снижению выхода ПЦР-продукта. На рисунке № 1.7. представленна электрофореграма с ПЦР продуктами, получиными при амплификации пети разных проб ДНК. Первые пять проб были получены при отжыге при 610С, вторые пять проб при 590С [57]. На данном рисунке видно как увеличение температуры приводит уменьшению выхода ПЦР-продукта, это может быть связано с: неполной гомологией праймеров, низким качеством ДНК-матрици и другими факторами.
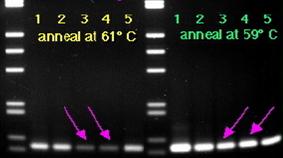
Рисунок №1.7.
На рисунке 1.8. изображена ситуация варьирования температуры плавления, количество амплифицируемых локусов и концентрации буфера которые изменяются в параллельных реакциях. Мы можем наблюдать много неспецифических полос при температуре отжига 48 °C, которые исчезают при увеличении температуры отжига. Хотя праймеры фланкирующие ПЦР продукт Y находятся во всех смесях в одинаковой концентрации самый сильный ПЦР продукт наблюдается в реакции амплификации с одной парой праймеров, затем идет мультиплексная ПЦР с пятью парами праймеров и самый слабый продукт Y наблюдался в мультиплексной ПЦР с семью парами праймеров. Двукратное увеличение концентрации ПЦР буфера позволяет сбалансировать выход легких продуктов но в тоже время приводит к увеличению выхода тяжелых не специфических продуктов.
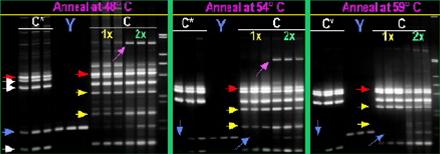
Рисунок № 1.8. Мультиплексная амплификация смеси С* (первые три дорожки каждого геля), пара праймеров “Y” (дорожки с 4 по 6) и смесь С (дорожки с 7 по 12 в однократном и двукратном ПЦР буфере) на базе трех разных ДНК матрицах, с использованием трех программ которые отличаются температуратурой отжига ((48° C, 54° C, 59° C). Дорожки 1-9 каждого геля демонстрируют ПЦР в однократном буфере. Дорожки 10-12 каждого геля демонстрируют ПЦР в двукратном буфере. На не помеченных дорожках нанесен маркер который содержит ПЦР-продукт длиной 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 т. п. н. Пять стрелок слева от первого геля показывают ожидаемый ПЦР-продукт смеси С (пять продуктов). Самый длинный специфический продукт на каждом геле маркирован красной стрелкой. Розовые стрелки указывают на сильный, тяжелый не специфический ПЦР продукт. Желтые стрелки указывают на два дополнительных продукта обусловленные смесью С. Синие стрелки указывают на продукт Y.
Отжиг происходит в соответствии с правилом комплементарности Чаргаффа, означающим, что в двухцепочечной молекуле ДНК напротив аденина всегда находится тимин, а напротив гуанина – цитозин (см. рис. 1.9). Если это условие не соблюдено, то отжига праймеров не происходит.
Рис. № 1.7. Принцип комплиментарности и антипараллельности цепей ДНК.
Определения температуры отжига.
В общем случае более высокая температура отжига обуславливает более высокую специфичность. Но как только температура отжига превышает некую критическую (для данной пары праймеров), количество продукта начинает резко снижаться.
Температура оптимального отжига праймеров зависит от:
- полимеразы;
- состава праймера (отношение CG к AT);
- последовательности праймера (nearest-neighbor взаимодействия);
- концентрации Na+ [61];
- концентрации праймера;
- концентрации Mg2+
- присутствия денатурирующих веществ таких как формамид, DMSO[58];
- последовательности между праймерами;
- pH;
- ионной силы ;
- концентрации ДНК;
- наличие других праймеров;
- наличие ингибиторов ПЦР;
Так как вышеперечисленные факторы учесть проблематично, то температуру отжига праймеров определяют эмпирически. При этом в качестве ориентира используют температуру плавления праймера. При использовании Tag-полимеразы рекомендуют использовать температуру отжига праймеров, рассчитанную согласно формуле (1.1). Фирма GENOSYS рекомендует использовать температуру отжига на 5-100С ниже температуры плавления[58]. Для Phusion™ Polymerase рекомендуется использовать температуру отжига на 30С больше температуры плавления при длине праймера более 20 п.н. Это объясняется тем, что Phusion™ Polymerase обладает способностью стабилизировать комплекс праймер-ДНК [48].
Tотжига = Tплавления - 50С (1.1)
Температура плавления - это температура, при которой половина нуклеиновых кислот находится в одноцепочечном состоянии, а вторая половина - в двухцепочечном.
Определения температуры плавления.
Для коротких ДНК олигонуклеотидов (14 – 20 пар оснований), при концентрации NaCl 0.9M рекомендуется уравнение (1.2).
Tплавления =2(#А+#Т)+4(#G+#С) (1.2)
где: #А, #Т, #G, #С – количество соответствующих нуклеотидов в последовательности праймера.
Для олигонуклеотидов длиной более 50 пар оснований при ![]() рекомендуется уравнение (1.3).
рекомендуется уравнение (1.3).
![]() (1.3)
(1.3)
где: ![]() - молярная концентрация моновалентного катиона (в данном случае
- молярная концентрация моновалентного катиона (в данном случае ![]() ),
), ![]() и
и ![]() - доля остатков G и C – в олигонуклеотиде, L – длина короткой цепи в дуплексе, F – процент формамида в растворе. Данное уравнение не применимо при
- доля остатков G и C – в олигонуклеотиде, L – длина короткой цепи в дуплексе, F – процент формамида в растворе. Данное уравнение не применимо при ![]() . [Wallace, R.M.; Israel, M.F.; Law, M.F.; Martin, M.A. J.Biol. Chem. 254, 4876(1979)]
. [Wallace, R.M.; Israel, M.F.; Law, M.F.; Martin, M.A. J.Biol. Chem. 254, 4876(1979)]
Однако эмпирически полученые уравнения (1.2) – (1.3) не согласуются с данными, приведенными в таблице № 1.7.
Таблица № 1.9. Вариирование последовательности олигонуклеотида при одинаковом составе [58].
|
№ пробы |
Последовательность(5'-3') |
|
M |
Длина |
GC |
|
1 |
AAAAACCCCCGGGGGTTTTT |
69.7 |
6103 |
20 |
50% |
|
2 |
ACGTACGTACGTACGTACGT |
57.2 |
6103 |
20 |
50% |
|
3 |
GATCGATCGATCGATCGATC |
64.5 |
6103 |
20 |
50% |
|
4 |
ATATATATATCGCGCGCGCG |
66.4 |
6103 |
20 |
50% |
Данные в таблице № 1.9. указывают на то, что температура плавления олигонуклеотида зависит не только от его длины и соотношения AT к CG, но и от расположения нуклеотидов в цепи. Это связано с тем, что нуклеотиды своими полярными и неполярными группами изменяют пространство вокруг себя и, таким образом, действуют на соседние нуклеотиды и на их водородные связи. Судя по таблице № 1.9. это действие достаточно велико. Для учета влияния последовательности нуклеотидов на устойчивость ДНК разработана модель, которая предполагает, что на каждый нуклеотид влияет два его соседа (Nearest Neighbor Model). Влияние каждого нуклеотида на устойчивость двухцепочечной ДНК оценивается по Энергии Гибса, а точнее по изменению энергии Гибса в реакции плавления двухцеточечной ДНК. Отрицательное значение энергии Гибса является движущей силой всех процессов известных науке. Если энергия Гибса равна нулю, то система находится в состоянии равновесия. Если энергия Гибса положительна, то протекает обратный процесс. Константа равновесия является отношением скорости прямого процесса (реакции) к скорости обратного процесса (реакции), и может быть получена из выражения (1.4).
![]() (1.4)
(1.4)
где: ΔG – изменение энергий Гибса в ходе реакции, T – температура, R – универсальная газовая постоянная. [51, 55, 66, 68].
Необходимо отметить, что, если концентрация одной из цепей ДНК взята в значительном избытке (как в случае присутствия праймеров), то константа равновесия будет равна равновесному соотношению двухцепочечных и одноцепочечных молекул ДНК. На рисунке № 1.10. представлена практически полученная зависимость доли двухцепочечных ДНК от температуры.
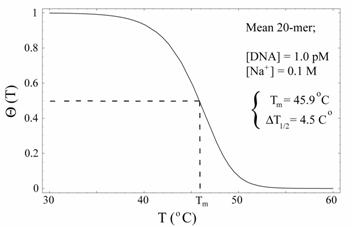
Рисунок № 1.10. Изменение отношения двухцепочечных ДНК ко всем молекулам ДНК от температуры [55].
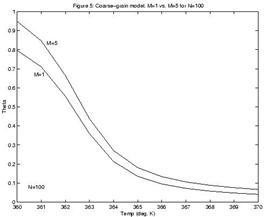
Рисунок № 1.11. Фракция двухцепочечной ДНК (Theta) как функция от температуры с различной долей АТ оснований. Расчет при помощи грубой апроксимации [54].
Разность энергий Гибса может быть определена из уравнения (1.5).
![]() (1.5)
(1.5)
где: ΔH – изменение энтальпии в ходе реакции,
ΔS - изменение энтропии в ходе реакции [51, 55, 59].
Изменение энтальпии и энтропии в ходе реакции образования комплекса ДНК-праймер может быть определено из принципа аддитивности, согласно которого ![]() ,
, ![]() [51]. Необходимо отметить, что принцип аддитивности может быть применен для рассчета изменения устойчивости комплекса праймер одноцепечечная ДНК в близи температцры плавления, но он требует коректировки энтальпии и энтропии. Необходимость коректировки энтальпии и энтропии связана с тем что энтальпия и энтропия являются функциями от температуры уравнение (1.6) и (1.7).
[51]. Необходимо отметить, что принцип аддитивности может быть применен для рассчета изменения устойчивости комплекса праймер одноцепечечная ДНК в близи температцры плавления, но он требует коректировки энтальпии и энтропии. Необходимость коректировки энтальпии и энтропии связана с тем что энтальпия и энтропия являются функциями от температуры уравнение (1.6) и (1.7).
![]() (1.6)
(1.6)
![]() (1.7)
(1.7)
где ![]() - изменение пеплоемкости в ходе реакции [60].
- изменение пеплоемкости в ходе реакции [60].
Влияние ![]() на температуру плавления описывается уравнением (1.8) [62].
на температуру плавления описывается уравнением (1.8) [62].
![]() (1.8)
(1.8)
где: R - универсальная газовая постоянная.
На рисунке № 1.11. представлена зависимость фракции двухцепочечной ДНК от температуры с различной долей АТ оснований расчет провел Lucas G. Nivon из Университета Гарварда [54]. Полученные данные близки к практическим (рис. № 1.10.). Однако принцип аддитивности не учитывает взаимодействий между соседними нуклеотидами. Что приводит к отсутствию зависимости между длиной двухцепочечного олигонуклеотида и температурой его плавления что противоречит практически полученным данным. Продемонстрируем это на простом примере. Допустим у нас есть ряд двухцепочечных олигонуклеотидов, состоящих из одинаковых нуклеотидов. Тогда ![]() ,
, ![]() и
и ![]() . Так как температура плавления - это та температура, при которой
. Так как температура плавления - это та температура, при которой ![]() , то
, то ![]() , что не согласуется с данными эксперимента, которые свидетельствуют о том, что чем больше олигонуклеотид, тем выше температура его плавления.
, что не согласуется с данными эксперимента, которые свидетельствуют о том, что чем больше олигонуклеотид, тем выше температура его плавления.
Вывод: существуют дополнительные взаимодействия, которые не учтены принципом аддитивности.
Для учета влияния последовательности нуклеотидов на устойчивость ДНК разработана модель, которая предполагает, что на каждый нуклеотид влияет два его соседа (Nearest Neighbor Model). На основании эмпирических исследований рассчитана энтальпия и энтропия для всех пар оснований, которые приведены в таблице № 1.10.
Таблица № 1.10. Термодинамические параметры для nearest-neighbor melting point formula.
|
Взаимодействие |
ДНК |
РНК |
||
|
ККал/моль |
ККал/К.моль |
ККал/моль |
ККал/К.моль |
|
|
AA/TT |
-9.1 |
-24.0 |
-6.6 |
-18.4 |
|
AT/TA |
-8.6 |
-23.9 |
-5.7 |
-15.5 |
|
TA/AT |
-6.0 |
-16.9 |
-8.1 |
-22.6 |
|
CA/GT |
-5.8 |
-12.9 |
-10.5 |
-27.8 |
|
GT/CA |
-6.5 |
-17.3 |
-10.2 |
-26.2 |
|
CT/GA |
-7.8 |
-20.8 |
-7.6 |
-19.2 |
|
GA/CT |
-5.6 |
-13.5 |
-13.3 |
-35.5 |
|
CG/GC |
-11.9 |
-27.8 |
-8.0 |
-19.4 |
|
GC/CG |
-11.1 |
-26.7 |
-14.2 |
-34.9 |
|
GG/CC |
-11.0 |
-26.6 |
-12.2 |
-29.7 |
|
Initiation |
0.0 |
-10.8 |
0.0 |
-10.8 |
Температура плавления олигонуклеотида согласно NN модели может быть рассчитана согласно уравнения (1.6).
![]() (1.6)
(1.6)
где: ![]() - сумарное изменение энтальпии, A – маленький, но черезвычайно важный коэфицент корректирующий образование двойной цепи,
- сумарное изменение энтальпии, A – маленький, но черезвычайно важный коэфицент корректирующий образование двойной цепи, ![]() - сумма изменения NN энтропии, R – универсальная газовая постоянная (1,99 кал/К.моль), С – концентрация олигонуклеотидов,
- сумма изменения NN энтропии, R – универсальная газовая постоянная (1,99 кал/К.моль), С – концентрация олигонуклеотидов, ![]() - концентрация катиона
- концентрация катиона ![]() .
.
После отжига праймеров Taq-полимераза начинает достраивание второй цепи ДНК с 3'-конца праймера.
2.3. Элонгация цепи.
На третьем этапе температуру в реакционной смеси доводят до оптимума работы Taq-полимеразы, и синтез второй цепи продолжается с максимальной эффективностью. Широко распространено использование Те= 72oС, т.к. на этой температуре полимеразная активность максимальна. Зависимость активности полимеразы от температуры приведена в таблице № 1.5. Активность определяется по включению метки в денатурированную и отожженную salmon sperm DNA, взятую в большом избытке.
Однако, Те= 72oС приемлема не для всех случаев:
1. В описании эксперимента для определения активности полимеразы есть важный момент: матрица взята в большом избытке, то есть, полимераза имеет возможность "пренебрегать" неудобными участками синтеза DNA. В реальном процессе полимеразе необходимо пройти все имеющиеся в наличии участки. Оказывается, что при температуре ~65oC синтез существенно меньше зависит от характера матрицы, что особенно актуально при синтезе GC-богатого фрагмента, как в FRAX – экспансии (но даже эта температура слишком велика для буфера с бетаином).
2. Матрицы с А-Т содержанием ~90% удаётся амплифицировать только снизив Те до температуры ~60oС (при этом скорость синтеза снижается до 1kb/min) [44].
Таблица № 1.11.
|
Температура oС |
Скорость синтеза |
|
|
нукл/сек |
kb/min |
|
|
75-80 |
~150 |
~9 |
|
70 |
>60 |
>3.6 |
|
55 |
24 |
1.4 |
|
37 |
1.5 |
190 |
|
22 |
0.25 |
15 |
В случае близкого значения температуры отжига праймеров и температуры оптимума работы фермента, становится возможным использовать двухэтапный ПЦР, совместив отжиг и элонгацию.
Температурный цикл амплификации многократно повторяется (30 и более раз). На каждом цикле количество синтезированных копий фрагмента ДНК удваивается.
Результатом циклического процесса является экспоненциальное увеличение количества специфического фрагмента ДНК, которое можно описать формулой (1.7)
![]() ~
~ ![]() , (1.7)
, (1.7)
где: А - количество специфических продуктов реакции амплификации; М – начальное количество ДНК-мишеней; n - число циклов амплификации.
Реальное значение эффективности отдельных циклов амплификации составляет около 78-97%. В случае присутствия в пробе ингибиторов реакции это значение может быть намного меньше, поэтому фактическое количество специфических продуктов амплификации лучше описывает уравнение (1.8).
![]() , (1.8)
, (1.8)
где: Е –эффективность реакции.
Следует заметить, что в процессе амплификации на исходной цепи синтезируются и длинные фрагменты, однако их накопление происходит лишь в арифметической прогрессии по формуле (1.9).
![]() , (1.9)
, (1.9)
где К – количество длинных продуктов амплификации.
Таким образом, специфические фрагменты, ограниченные на концах праймерами, впервые появляются в конце второго цикла, накапливаются в геометрической прогрессии и очень скоро начинают доминировать среди продуктов амплификации.
3. Пост-PCR.
Циклы после PCR проводятся для превращения некоторого остаточного количества одноцепочечных матриц (подвижность на электрофорезе ниже) в двухцепочечные (более прямой способ решить эту задачу - провести несколько дополнительных (2-5) циклов без денатурации: только отжиг и удлинение);
1.7.3. Число циклов амплификации.
Чем больше число ПЦР-циклов, тем выше чувствительность метода и ниже специфичность. Однако максимальной чувствительности нельзя достичь только увеличением числа циклов, необходимо оптимизировать и другие параметры, с тем чтобы повысить эффективность каждого цикла. Число циклов амплификации зависит также от содержания в образце нужной последовательности. Если ставится цель выявления уникальных генов, присутствующих в каждой клетке (например, при ПЦР-диагностике наследственных заболеваний), то достаточно 20—30 циклов, если же последовательность-мишень имеется только в части клеток исследуемого препарата (например, в случае вирусной инфекции или в опухолях), то число циклов может составлять 30 - 50.
Необходимо отметить, что при увеличении числа циклов выход продукта проходит три фазы:
1. Выход продукта увеличивается в геометрической прогресии
2. Выход продукта стабилизируется в следствии «Эффекта плато», который рассмотрен в п. 1.7.2.4.
3. Снижение концентрации ПЦР-продукта [57].
На рисунке № 1.11. приведен снимок электрофореза ПЦР продуктов, которые были получены при амплификации двух образцов ДНК с варьированием числа циклов амплификации от 21 до 45, с шагом 3 цикла [57].
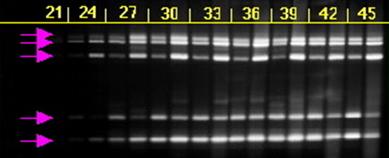
Рисунок № 1.11.
1.7.4. Эффект плато при увеличении количества ПЦР циклов.
Следует заметить, что процесс накопления специфических продуктов амплификации по геометрической прогрессии идет лишь ограниченное время, а затем его эффективность критически падает. Это связано с так называемым «эффектом плато».
Термин «эффект плато» используют для описания процесса накопления продуктов ПЦР на последних циклах амплификации, когда количество ампликонов достигает 0,3 - 1 пмолей.
В зависимости от условий и количества циклов реакции амплификации, на момент достижения «эффекта плато» влияют:
- утилизация субстратов (дНТФ и праймеров);
- стабильность реагентов (дНТФ и фермента);
- количество ингибиторов, включая пирофосфаты и ДНК-дуплексы;
- неспецифические продукты или праймер-димеры, конкурирующие за праймеры, дНТФ и полимеразу;
- концентрация специфического продукта;
- неполная денатурация при высокой концентрации продуктов амплификации.
Чем меньше начальная концентрация ДНК-мишени, тем выше риск выхода реакции на плато. Этот момент может наступить до того, как количество специфических продуктов амплификации будет достаточно, чтобы их можно было проанализировать. Избежать этого позволяют лишь хорошо оптимизированные тест-системы.
1.7.5. Способы постановки ПЦР
1.7.5.1. Способ постановки ПЦР с использованием «горячего старта»
Чтобы уменьшить риск образования неспецифических продуктов реакции амплификации, используют подход, получивший название «горячий старт» (от англ. «hot-start»). Суть его состоит в предотвращении возможности начала реакции до момента достижения в пробирке условий, обеспечивающих специфический отжиг праймеров.
Дело в том, что в зависимости от ГЦ-состава и размера, праймеры имеют определенную температуру плавления (Tm), при которой образование водородных связей нестабильно. Если температура системы превышает Тm, праймер не в состоянии удерживаться на цепи ДНК и денатурирует. При соблюдении оптимальных условий, т.е. температуры отжига близкой к температуре плавления, праймер образует двухцепочечную молекулу только при условии его полной комплементарности и, таким образом, обеспечивает специфичность реакции.
Существуют различные варианты реализации «горячего старта»:
1. внесение в реакционную смесь Taq-полимеразы во время первого цикла после прогрева пробирки до температуры денатурации;
2. разделение ингредиентов реакционной смеси прослойкой, например, парафина или воска на части (в нижней - праймеры, в верхней - Taq-полимераза и ДНК-мишени), которые смешиваются при достижении температуры плавления материала прослойки (~45-85ºС);
3. использование моноклональных антител к Taq-полимеразе. Фермент, связанный моноклональными антителами, становится активным лишь после стадии первой денатурации, когда моноклональные антитела необратимо денатурируют, и освобождают активные центры Taq-полимеразы.
Во всех перечисленных случаях, даже если неспецифический отжиг произошел до начала температурного циклирования, элонгации не происходит, а при нагревании комплексы праймер-ДНК денатурируют, поэтому неспецифические продукты не образуются. В дальнейшем температура в пробирке не опускается ниже температуры плавления, что обеспечивает образование специфического продукта амплификации.
1.7.5.2. «Nested» ПЦР
Одним из способов повысить чувствительность реакции является применение метода «nested» (гнездной) амплификации. Суть его заключается в последовательном использовании двух пар праймеров – внешней и внутренней. После использования первой пары праймеров продукт амплификации переносят в другую пробирку с внутренней парой праймеров. Иногда вместо «nested» амплификации используют процесс реамплификации. В этом случае проводят дополнительный раунд амплификации, в котором используют прежнюю пару праймеров, а в качестве ДНК-мишени – продукт первой реакции амплификации.
Такая схема постановки амплификации более трудоемка, поскольку, как правило, делает необходимым постановку двух реакций амплификации вместо одной и требует особенно тщательного обустройства лабораторных помещений, позволяющих гарантированно избегать контаминации продуктами реакции после использования внешней пары праймеров. К «гнездной» амплификации или реамплификации прибегают лишь в особых случаях, так как современные ПЦР-наборы позволяют добиваться тех же результатов иными средствами.
1.7.5.3. Полуколичественный анализ
Для более точной оценки количества ДНК-мишени в реакционной смеси предпринимаются специальные подходы, известные под общим названием полуколичественного ПЦР-анализа. Приставка «полу» имеет принципиальное значение из-за условной точности результатов этого анализа.
К таким подходам следует отнести использование специального приборного обеспечения в сочетании с препаратами специфической ДНК с известной концентрацией.
К приборному обеспечению, позволяющему получать полуколичественные результаты ПЦР анализа, следует отнести модели «iCycler IQtm» (Био-Рад, США) и «COBAS Amplicor» (Roche, США), которые позволяют следить за кинетикой накопления продуктов амплификации. Это достигается красивым решением на стыке физики и биологии. В реакционную смесь вводят гибридизационные зонды, в состав которых входят нуклеотиды, меченные особыми реактивами – флуорофором и тушителем. Флуорофоры способны излучать энергию лишь в свободном от тушителя состоянии. На стадии отжига происходит гибридизация зондов с внутренними участками ампликонов. В процессе элонгации Taq- полимераза разрушает зонды за счет своей экзонуклеазной активности. Это приводит к попаданию меченых нуклеотидов в раствор, где они начинают флуоресцировать (рис. 1.12.). Интенсивность флуоресценции фиксируется специальным детектором и пропорциональна количеству продуктов амплификации. Если гибридизация не проходит, то зонд остается целым, а флуорофоры, входящие в его состав, не дают излучения.
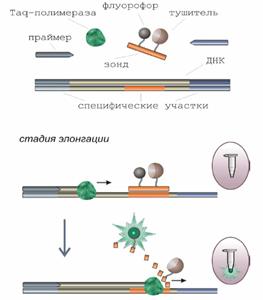
Рис. 1.12. Использование флаоресцентных зондов в троцессе амплификации.
При исследовании образцов каждая серия экспериментов сопровождается постановкой амплификации с контрольными образцами, в которых заведомо известно количество копий ДНК. Сравнение кинетики накопления продуктов амплификации в реакционных смесях в экспериментальном и контрольных образцах позволяет полуколичественно оценить концентрацию ДНК в диапазоне разведений контрольных препаратов ДНК.
Более простым, но менее надежным вариантом оценки количества специфической ДНК является метод разведений. Детекцию проводят после гибридизации с ДНК-зондами, или методом электрофореза, определяя количество работающих разведений.
При всей привлекательности полуколичественного анализа следует отметить, что для его выполнения допускается использование только препаратов ДНК с высокой степенью очистки. Важно отметить, что существуют примеси, которые могут по-разному влиять на эффективность амплификации исследуемой и контрольной ДНК. При использовании упрощенных методов выделения ДНК в большинстве случаев не представляется возможным заранее предсказать количество и состав примесей в клинических образцах.
Известно, что в некоторых случаях возможны потери ДНК на стадии выделения, что может существенно исказить значение реального количества ДНК в образце.
Кроме того, точное знание количества возбудителей в конкретном клиническом образце далеко не всегда может дать полное представление об инфекционном процессе, например, из-за различной локализации и вирулентности микроорганизмов, а также состояния иммунной системы организма хозяина.
Таким образом, существующие варианты полуколичественного анализа пока еще не имеют большой практической ценности для рутинного клинического тестирования. В то же время их польза неоспорима при оценке эффективности терапии при некоторых инфекционных заболеваниях.
1.7.6. Контроль за прохождением реакции амплификации
Результат ПЦР можно квалифицировать как положительный или отрицательный в зависимости от того, обнаружена в образце интересующая нас последовательность-мишень или нет. Однако нарушение нормального хода амплификации, недостаточная чувствительность метода и непредвиденный полиморфизм последовательности-мишени в области связывания праймеров или гибридизационного зонда может дать ложноотрицательный результат. В случае загрязнения образцов и случайной гомологии между зондом, праймерами и последовательностью, сходной с мишенью, получаются ложноположительные результаты. Проблемы, которые возникают в связи с перекрестными реакциями с участием зонда или праймеров и возможным полиморфизмом.
Очень важным для правильной интерпретации результатов является выбор контролей. Положительные и отрицательные контроли должны быть хорошо охарактеризованы. Часто используют ДНК из клеточных линий, заведомо содержащих или не содержащих последовательность-мишень. В каждом анализе нужны как минимум три контроля:
• положительный контроль;
• отрицательный контроль;
• бланк-контроль.
Бланк-контроль - это реакционная смесь, в которой присутствуют все компоненты, за исключением ДНК; он является индикатором загрязнений. Один тип положительного контроля должен содержать максимальное число последовательностей-мишеней, другой — небольшое их число. Это позволяет определить чувствительность и эффективность ПЦР.
В некоторых случаях амплификация вообще не идет, и если есть основания думать, что полученный результат ложноотрицательный, очень трудно определить, по какой именно причине. Чтобы решить эту проблему, каждый образец необходимо тестировать, проведя амплификацию какой-нибудь геномной последовательности, например гена рецептора липопротеинов низкой плотности или ß-глобинового гена. При этом размер геномной последовательности-мишени должен быть немного больше, чем исследуемой; это позволит убедиться, что ДНК образца не слишком сильно деградирована. При ПЦР любого образца тканей человека геномная ДНК должна амплифицироваться. Отсутствие амплификации может означать ингибирование фермента, сильную деградацию ДНК или то, что ее слишком мало. В этом случае надо повторить амплификацию образца, увеличив и уменьшив количество ДНК-матрицы или увеличив концентрацию ДНК-полимеразы Tag. Амплификация может отсутствовать несмотря на все предпринятые усилия. Это может быть связано с сильным некрозом исследуемой ткани или фиксацией препарата не в 10% формалине, а в других фиксаторах. В этом случае единственным выходом является повторная биопсия.
Геномные праймеры можно использовать в одном раунде ПЦР вместе с праймерами, специфичными в отношении последовательности-мишени: амплификация двух последовательностей идет при этом одновременно. Такой множественный ПЦР-тест может быть менее чувствительным, чем тот, в котором используется один набор праймеров, но он исключает появление ложноотрицательного результата.
1.8. Выдиление ДНК матрици
При подготовке пробы к постановке ПЦР используют различные методики в зависимости от поставленных задач. Их суть заключается в экстракции (извлечении) ДНК из биопрепарата и удалении или нейтрализации посторонних примесей для получения препарата ДНК с чистотой, пригодной для постановки реакции амплификации.
Иногда бывает достаточно прокипятить образец в течение 5-10 минут, однако в большинстве случаев требуются более трудоемкие методы.
Стандартной и ставшей уже классической считается методика получения чистого препарата ДНК, предложенная Marmur. Она включает в себя ферментативный протеолиз клеток с последующей депротеинизацией и переосаждением ДНК спиртом. Однако это метод довольно трудоемок и предполагает работу с такими агрессивными и имеющими резкий запах веществами, как фенол и хлороформ.
Одним из популярных в настоящее время является метод выделения ДНК, предложенный Boom с соавторами. Этот метод основан на использовании для лизиса клеток сильного хаотропного агента - гуанидина тиоционата (GuSCN) и последующей сорбции ДНК на носителе (стеклянные бусы, диатомовая земля, стеклянное «молоко» и т.д.). После отмывок в пробе остается ДНК, сорбированная на носителе, с которого она легко снимается с помощью элюирующего буфера. Метод удобен, технологичен и пригоден для подготовки образца к амплификации. Однако возможны потери ДНК вследствие необратимой сорбции на носителе, а также в процессе многочисленных очисток. Особенно большое значение это имеет при работе с небольшими количествами ДНК в образце. Кроме того, даже следовые количества GuSCN могут ингибировать ПЦР. Поэтому при использовании этого метода очень важен правильный выбор сорбента и тщательное соблюдение технологических нюансов. Следует отметить, что из-за большого количества стадий добавления и удаления растворов при работе с образцом требуется аккуратность, т.к. возможна перекрестная контаминация между пробами образующейся аэрозолью ДНК.
Другая группа методов пробоподготовки основана на использовании ионообменников типа Chilex, которые, в отличие от стекла, сорбируют не ДНК, а, наоборот, примеси, мешающие реакции. Как правило, эта технология включает две стадии: кипячение образца, в результате чего клеточные стенки разрушаются, а нуклеиновые кислоты выходят в раствор; и сорбция примесей на ионообменнике. Метод чрезвычайно привлекателен простотой исполнения. В большинстве случаев он пригоден для работы с клиническим материалом. К сожалению, иногда встречаются образцы с такими примесями, которые невозможно удалить с помощью ионообменников. Кроме того, клеточные стенки некоторых микроорганизмов не поддаются разрушению простым кипячением. В этих случаях необходимо введение дополнительных стадий обработки образца.
При массовом скрининге, когда важно получить статистические данные, допускается использование простых методов с применением детергентов или обработки биологического материала щелочами с последующей их нейтрализацией. В то же время, использование подобных методов пробоподготовки для клинической диагностики может приводить к ложноотрицательным результатам вследствие использования в реакционной смеси некачественного препарата ДНК.
Таким образом, к выбору метода пробоподготовки следует относиться с пониманием целей проведения предполагаемых анализов.
1.9. Детекция
Для правильной оценки результатов ПЦР важно понимать, что данный метод не является количественным. Теоретически продукты амплификации единичных молекул ДНК-мишени могут быть обнаружены с помощью электрофореза уже после 30-35 циклов. Однако на практике это выполняется лишь в случаях, когда реакция проходит в условиях, близких к идеальным, что в жизни встречается не часто. Особенно большое влияние на эффективность амплификации оказывает степень чистоты препарата ДНК, т.е. наличие в реакционной смеси тех или иных ингибиторов, от которых избавиться в некоторых случаях бывает крайне сложно. Иногда из-за их присутствия не удается амплифицировать даже десятки тысяч молекул ДНК-мишени. Таким образом, прямая связь между исходным количеством ДНК-мишени и конечным количеством продуктов амплификации часто отсутствует.
1.9.1. Метод горизонтального электрофореза
Для визуализации результатов амплификации используют различные методы. Наиболее распространенным на сегодняшний день является метод электрофореза, основанный на разделении молекул ДНК по размеру. Для этого готовят пластину агарозного геля, представляющего собой застывшую после расплавления в электрофорезном буфере агарозу в концентрации 1,5-2,5% с добавлением специального красителя ДНК, - например, бромистого этидия. Застывшая агароза образует пространственную решетку. При заливке с помощью гребенок в геле формируют специальные лунки, в которые в дальнейшем вносят продукты амплификации. Пластину геля помещают в аппарат для горизонтального гель-электрофореза и подключают источник постоянного напряжения. Отрицательно заряженная ДНК начинает двигаться в геле от минуса к плюсу. При этом более короткие молекулы ДНК движутся быстрее, чем длинные. На скорость движения ДНК в геле влияет концентрация агарозы, напряженность электрического поля, температура, состав электрофорезного буфера и, в меньшей степени, ГЦ-состав ДНК. Все молекулы одного размера движутся с одинаковой скоростью. Краситель встраивается (интеркалирует) плоскостными группами в молекулы ДНК. После окончания электрофореза, продолжающегося от 10 мин до 1 часа, гель помещают на фильтр трансиллюминатора, излучающего свет в ультрафиолетовом диапазоне (254, 310 нм). Энергия ультрафиолета, поглощаемая ДНК в области 260 нм, передается на краситель, заставляя его флуоресцировать в оранжево-красной области видимого спектра (590 нм).
Яркость полос продуктов амплификации может быть различной. Поэтому часто в ПЦР-лабораториях принято оценивать результат по трех-, четырех- или пятибалльной системе. Однако, как уже отмечалось ранее, это нельзя связывать с начальным количеством ДНК-мишени в образце. Часто уменьшение яркости свечения полос связано со снижением эффективности амплификации под влиянием ингибиторов или других факторов.
1.9.2. Метод вертикального электрофореза
Метод вертикального электрофореза принципиально схож с горизонтальным электрофорезом. Их отличие заключается в том, что в данном случае вместо агарозы используют полиакриламид. Его проводят в специальной камере для вертикального электрофореза. Электрофорез в полиакриламидном геле имеет большую разрешающую способность по сравнению с агарозным электрофорезом и позволяет различать молекулы ДНК разных размеров с точностью до одного нуклеотида. Приготовление полиакриламидного геля несколько сложнее агарозного. Кроме того, акриламид является токсичным веществом. Поскольку необходимость определить размер продукта амплификации с точностью до 1 нуклеотида возникает редко, то в рутинной работе этот метод не используют.
1.10. Проблема контаминации
Потенциально высокая чувствительность полимеразной цепной реакции делает совершенно необходимым особенно тщательное устройство ПЦР-лаборатории. Это связано с наиболее острой проблемой метода – контаминацией.
Контаминация – попадание из внешней среды в реакционную смесь специфических и неспецифических молекул ДНК, способных служить мишенями в реакции амплификации и давать ложноположительные или ложноотрицательные результаты.
Такими мишенями могут быть продукты реакции, попадающие во внешнюю среду на этапе детекции из пробирок, в которых успешно прошла амплификация, либо специфическая ДНК из образцов на этапе пробоподготовки.
Существует несколько способов борьбы с этим неприятным явлением. Одним из них является использование фермента N-урацил-гликозилазы (УГ). В основе этого метода лежит способность УГ расщеплять молекулы ДНК со встроенным урацилом. Реакцию амплификации проводят с использованием смеси дНТФ, в которой дТТФ заменен на дУТФ (урацил), и после термоциклирования все образующиеся в пробирке ампликоны будут содержать урацил. Если до амплификации в реакционную смесь добавить УГ, то попавшие в реакционную смесь ампликоны будут разрушены, тогда как нативная ДНК останется целой и будет в дальнейшем служить мишенью для амплификации.
Другим способом инактивации ампликонов служит фотохимическое воздействие на молекулы ДНК. Для этого используют псорален или изопсорален, которые активируются кратковременным облучением ультрафиолетовым светом. Модифицированные этими соединениями молекулы ДНК не могут участвовать в реакции амплификации.
Однако, как известно, ни одна биологическая или химическая реакция не идёт со 100% эффективностью и, соответственно, после инактивации продуктов амплификации из миллиардов копий амплифицированного фрагмента хотя бы несколько останутся целыми, что существенно снижает ценность такого подхода. Кроме того, всегда остается риск кросс-контаминации от образца к образцу в процессе пробоподготовки.
Таким образом, оба эти метода лишь в некоторой степени позволяют устранить источник контаминации и не гарантируют от ложноположительных и ложноотрицательных результатов.
Для уверенности в отсутствии контаминации необходимо каждую серию экспериментов сопровождать отрицательными контролями. В качестве отрицательных контролей рекомендуется использовать воду из комнаты пробоподготовки (после каждого десятого клинического образца желательно обрабатывать вместо биологического образца пробирку с водой).
Все реактивы рекомендуется хранить разлитыми на отдельные порции (аликвоты).
Если все же, несмотря на принятые меры, обнаружены следы контаминации, то все используемые порции реактивов рекомендуется заменить на новые, а все поверхности помещения, оборудования, пипеток и пр. обработать 0,1 М HCl, либо хлорсодержащими препаратами, например, ДП-2Т.
