на магістерську атестаційну роботу
Островного Дениса Володимировича
1. Тема роботи: “Використання методів ДНК-аналізу для діагностики моногенних спадкових захворювань” затверждена наказом по університету від “ ” 2003 р.
2. Мета дослідження - провести ДНК-аналіз мутацій та поліморфізму гена ФАГ і оптимізувати технологію проведення полімеразної ланцюгової реакції для ампліфікації in vitro послідовності ДНК сьомого екзона гена ФАГ.
3. Вихідні дані: звіти з НДР
4. Основні задачі дослідження:
4.1. ДНК-аналіз мутацій R408W, IVS12nt1, R158Q, Y414C, IVS10nt546 гену ФАГ
4.2. ДНК-аналіз мутацій в послідовності 7-го екзона гена ФАГ
4.3. Аналіз алельного поліморфізму VNTR-локуса 3`-нетрансльованої області гена ФАГ
5. Термін подання роботи до захисту _________________
6. Дата видачі завдання 25.10. 2003
|
Науковий керівник |
________________ підпис |
|
Завдання прийняв до виконання |
________________ підпис |
ЗМІСТ
|
сторінка |
||
|
Перелік умовних скорочень |
5 |
|
|
Вступ |
6 |
|
1. Огляд літератури |
8 |
|
1.1. |
Історія дослідження природи фенілкетонурії |
8 |
|
|
1.2. |
Картування і клонування гена фенілаланінгідроксилази |
11 |
|
|
1.3. |
Структура гена фенілаланінгідроксилази |
12 |
|
|
1.4. |
Білковий продукт гена ФАГ |
13 |
|
|
1.5. |
Поліморфні послідовності ДНК гена ФАГ |
14 |
|
|
1.6. |
Мутації гена фенілаланінгідроксилази |
17 |
|
|
1.7. |
Полімеразна ланцюгова реакція |
19 |
|
1.7.1. |
Компоненти ампліфікаційної реакційної суміші |
21 |
|
1.7.2. |
Температурний режим ампліфікації |
32 |
|
1.7.3. |
Кількість циклів ампліфікації |
46 |
|
1.7.4. |
Ефект плато при збільшенні кількості ПЛР циклів |
47 |
|
1.7.5. |
Модифікації постановки ПЛР |
48 |
|
1.7.6. |
Контроль за проходженням реакції ампліфікації |
52 |
|
1.8. |
Виділення ДНК матриці |
54 |
|
1.9. |
Детекція продукту полімеразної ланцюгової реакції |
56 |
|
|
1.10. |
Проблема контамінації |
58 |
|
2. Експериментальна частина. |
60 |
|
|
2.1. |
Об'єкти та методи досліджень |
60 |
|
2.1.1. |
Матеріали досліджень |
60 |
|
2.1.2. |
Методи досліджень |
62 |
|
2.1.2.1. |
Виділення й очищення ДНК |
62 |
|
2.1.2.2. |
Ампліфікація послідовностей гена ФАГ методом полімеразної ланцюгової реакції |
64 |
|
2.1.2.3. |
Ідентифікація мутацій методом рестрикційного аналізу |
66 |
|
2.1.2.4. |
69 |
|
|
2.2. |
Результати та їх обговорення |
70 |
|
2.2.1. |
ДНК-аналіз мутацій R408W, IVS12nt1, R158Q, Y414C, IVS10nt546 гена ФАГ |
70 |
|
2.2.2. |
ДНК-аналіз мутацій у послідовності 7-го екзона гена ФАГ |
76 |
|
2.2.3. |
Аналіз алельного поліморфізму VNTR-локусу 3`-нетрансльованої області гена ФАГ |
80 |
|
2.2.4. |
Оптимізація умов проведення ампліфікації |
81 |
|
2.2.4.1. |
Специфічність відпалу праймерів |
81 |
|
2.2.4.2. |
Постановка задачі оптимізації |
83 |
|
2.2.4.3. |
Отримання математичної моделі ампліфікації |
84 |
|
3. Висновки. |
112 |
|
|
Перелік посилань |
113 |
|
|
Додатки |
121 |
|
Перелік умовних скорочень
|
БСА |
- |
бичачий сироватковий альбумін |
|
ДНК |
- |
дезоксирибонуклеїнова кислота |
|
ЕДТА |
- |
етилендіамінтетраамінуксусна кислота |
|
ПААГ |
- |
поліакриламідний гель |
|
ПДРФ |
- |
поліморфізм довжини рестрикційних фрагментів |
|
ПЛР |
- |
полімеразна ланцюгова реакція |
|
РНК |
- |
рибонуклеїнова кислота |
|
ФАГ |
- |
фенілаланінгідроксилаза |
|
ФКУ |
- |
фенілкетонурія |
|
ФПК |
- |
фенілпіровиноградна кислота |
|
ssDNA |
- |
однолакцюгова дезоксирибонуклеїнова кислота |
|
ДНК.Пр, DNA.Pr |
- |
комплекс однолакцюгової дезоксирибонуклеїнової кислоти з праймером |
|
dNTP, дНТФ |
- |
дезоксінуклеотидтрифосфат |
|
STR |
- |
короткі тандемні повтори (short tandeme repeat) |
|
VNTR |
- |
варіююча кількість тандемних повторів, (variable number tandem repeat) |
|
п.н. |
- |
пар нуклеотидів |
|
т. п. н., kb |
- |
тисяч пар нуклеотидів |
|
kD |
- |
кілодальтон |
|
Tris |
трис-(оксиметил)-амінометан tris(hydroxymethyl)aminomethane C4H11O3N |
|
|
NN |
- |
Nearest Neighbor |
Вступ
У зв'язку з розшифровкою послідовності ДНК генома людини на передній план виходить питання з'ясування функціональної ролі генів. Найбільш перспективним підходом у вивченні генів людини, в умовах, коли проведення експериментів обмежене особливостями досліджуваного об'єкта, є аналіз генів спадкових захворювань. Вже до 2000 року на хромосомах людини картовано близько 1000 генів, мутації в яких призводять до спадкових захворювань [1]. Одним з перших у геномі людини був картований і ідентифікований ген фенілаланінгідроксилази, мутації в якому обумовлюють розвиток захворювання з аутосомно-рецессивним типом спадкування – фенілкетонурії [2]. Вивчення молекулярно-генетичної природи цього захворювання дозволяє досліджувати частоту, природу і механізми поширення мутацій у геномі людини. Аналіз асоціації генотипу і клінічних проявів захворювання відбиває фенотиповий ефект генетичних змін, а також є основою для перспективних досліджень функціонування і взаємодії генів.
З практичної точки зору, дослідження молекулярно-генетичних основ спадкових хвороб людини відкриває нові можливості в діагностиці, профілактиці і лікуванні важких патологій спадкоємної природи. Реальністю сьогоднішнього дня є застосування програм генетичного тестування і пренатальної ДНК-діагностики, багатьох захворювань, а справою найближчого майбутнього – впровадження в клінічну практику методів генотерапії.
Мета і задачі дослідження. Мета роботи - провести ДНК-аналіз мутацій та поліморфізму гена ФАГ і оптимізувати технологію проведення полімеразної ланцюгової реакції для ампліфікації in vitro послідовності ДНК сьомого екзона цього гена.
Для досягнення мети були поставлені основні задачі дослідження:
1. ДНК-аналіз мутацій R408W, IVS12nt1, R158Q, Y414C, IVS10nt546 гену ФАГ
2. ДНК-аналіз мутацій в послідовності 7-го екзона гена ФАГ
3. Аналіз алельного поліморфізму VNTR-локуса 3`-нетрансльованої області гена ФАГ
Об'єктом дослідження є ДНК людини, отримана з лейкоцитів периферійної крові, а предметом – методи аналізу мутацій у 5, 7, 12 екзоні та 10, 12 інтроні гена ФАГ.
У роботі використовувалися методи: виділення й очищення ДНК, ампліфікація in vitro послідовності гену ФАГ методом полімеразної ланцюгової реакції, рестрикційний аналіз, гель-електрофорез.
Наукова новизна отриманих результатів.
Була розроблена математична модель відпалу праймерів, яка дозволила отримати оптимальний температурний режим ампліфікації.
Практичне значення отриманих результатів. Була розроблена оптимізована методика для діагностики мутацій, локалізованих у послідовності сьомого екзону гена ФКУ.
Розроблено стратегію використання прямого аналізу мутацій гена ФАГ, а також аналіз зчеплення мутантного гена з високоінформативними ДНК-поліморфними маркерами для пренатальної діагностики, масового і селективного скринінгу гетерозиготних носіїв мутантних генів з метою профілактики цього важкого спадкового захворювання в Україні.
Автор висловлює щиру вдячність співробітникам відділу генетики людини ІМБІГ НАН України за постійну підтримку і допомогу під час виконання роботи.
1. Огляд літератури
1.1. Історія дослідження природи фенілкетонурії
Фенілкетонурія – одне з найбільш розповсюджених спадкових захворювань групи ензимопатій моногенної природи з аутосомно-рецессивним типом спадкування.
Вперше в медичній літературі опис цього захворювання з'явився в 1934 році. [3]
Клінічні симптоми фенілкетонурії з'являються вже в перші місяці життя і можуть бути розподілені на такі групи: інтелектуальний дефект – розумова відсталість (частіше в ступені імбіцильності або глибокої дебільності); нейродегенеративні прояви; судорожний синдром [4].
Серед інших симптомів виділяють: косоокість, що сходиться і розходиться, асиметрія обличчя, зниження глоткового і піднебінного рефлексів; вегетативно-трофічні розлади: сухість шкіри, порушення її пігментації, ламкість волосся, тенденція до артеріальної гіпотонії [5].
Основною діагностичною ознакою захворювання є підвищена концентрація фенілаланіну в крові, що визначалася раніше методом Гатри, а тепер методом імунохімічного аналізу. В нормі у дітей цей показник складає 62±18 ![]() M/л, а у хворих на ФКУ - 120- 2400
M/л, а у хворих на ФКУ - 120- 2400 ![]() M/л [1]. У більшості країн світу розроблені програми скринінгу немовлят на фенілкетонурію. В Україні ця програма діє, починаючи з 1971року. Завдяки цим програмам не тільки стало можливим починати пресимптоматичне лікування хворих, але і визначати частоту цього захворювання в різних країнах. Так максимальна частота зареєстрована в Ірландії 1:4500, а мінімальна в Японії 1:120000 [6, 7]. В Україні на 8300 немовлят народжується одна дитина хвора на ФКУ [8], причому в різних регіонах частота захворювань на ФКУ істотно відрізняється. Наприклад, у Криворізькій області - 1:12000, а у Полтавській і Чернігівській областях - 1:4419. Частота носіїв гена ФКУ в Українській популяції 1:43 [9].
M/л [1]. У більшості країн світу розроблені програми скринінгу немовлят на фенілкетонурію. В Україні ця програма діє, починаючи з 1971року. Завдяки цим програмам не тільки стало можливим починати пресимптоматичне лікування хворих, але і визначати частоту цього захворювання в різних країнах. Так максимальна частота зареєстрована в Ірландії 1:4500, а мінімальна в Японії 1:120000 [6, 7]. В Україні на 8300 немовлят народжується одна дитина хвора на ФКУ [8], причому в різних регіонах частота захворювань на ФКУ істотно відрізняється. Наприклад, у Криворізькій області - 1:12000, а у Полтавській і Чернігівській областях - 1:4419. Частота носіїв гена ФКУ в Українській популяції 1:43 [9].
L-фенілаланін належить до числа незамінних амінокислот. У нормальних умовах лише незначна його частина використовується для побудови власних білків, а більша частина перетворюється в тирозин [4].
У 1953 році Джервис і співавтори показали, що в хворих на фенілкетонурію перетворення фенілаланіну в тирозин майже не відбувається [10]. Пізніше було встановлено, що причиною цього є відсутність або значне зниження активності лабільної (печіночної) фракції ферменту фенілаланінгідроксилази [11]. Внаслідок цього концентрація фенілаланіну в усіх рідких середовищах організму зростає в десятки разів. На противагу фенілаланіну концентрація тирозину знижується [12].
Реакція перетворення фенілаланіну в тирозин здійснюється спеціальною ферментативною системою - гідроксилази L-фенілаланіна (ФАГ, фенілаланін 4-монооксигеназа, ЄС 1.14.16.1), що каталізує приєднання гідроксильної групи до фенілаланіну. До складу ферменту входять два компоненти: лабільний і стабільний [13, 14]. Стабільна фракція синтезується в багатьох органах, у тому числі нирках, серці, печінці; лабільна фракція синтезується тільки в печінці [5]. У реакції перетворення фенілаланіну в тирозин беруть участь кисень і тетрагідробіоптерин, що виступає в ролі кофактора, а також дегідроптеридинредуктаза (ДГПР, КФ 1.6.99. 7) [13, 14, 15, 16, 17]. Реакція відбувається за наступною схемою:
фенілаланін + О2 + тетрагідробіоптерин![]() тирозин + вода + окислений біоптерин [18].
тирозин + вода + окислений біоптерин [18].
Ця реакція відбувається при участі лабільної фракції фенілаланінгідроксилази. Стабільна частина ферментативної системи, а також ДГПР виконують функцію відновника окисленого біоптерина в тетрагідробіоптерин [14].
У хворих фенілкетонурією значна частина фенілаланіну виводиться з організму із сечею, крім того, для зниження концентрації фенілаланіну відбувається активація побічного шляху перетворення амінокислоти – переамінирування. Близько 10 % фенілаланіну, що надходить з їжею, у хворих фенілкетонурією виділяється із сечею у вигляді фенілаланіну, 30 % - у вигляді фенілпіровиноградної кислоти (ФПК), 20 % - фенілмолочної кислоти і 10 % - фенілуксусної кислоти [19].
Первинний дефект синтезу фенілаланінгідроксилази приводить до нагромадження в організмі людини фенілаланіну і його похідних. Наслідком цього є інгібірування ряду ферментів, що спричиняє вторинні порушення обміну важливих амінокислот, у першу чергу тирозину і триптофану [4].
Тирозин бере участь у будівництві надзвичайно важливих білків організму, а саме меланіну, норадреналіна, адреналіну, тироксину, дофаміна [20, 21]. На перший погляд видно, що дефіциту тирозину, і, як наслідок, речовин, що з нього виходять, при ФКУ не повинне бути, оскільки тирозин надходить у організм із їжи. Однак фенілаланін і його аномальні метаболіти інгібірують ферменти, що беруть участь в обміні тирозину і триптофану [21].
1.2. Картування і клонування гена фенілаланінгідроксилази
Роботи з картування гена ФАГ були початі ще в 70-і роки. Спочатку картування проводилося на основі аналізу зчеплення з іншими поліморфними білковими маркерами людини. Було показано, що ген ФАГ зчеплений з геном PGM-1 (фосфоглюкомутаза), а також з геном AMY-1 AMY-2 (амілазний локус) картований на першій хромосомі [22, 23]. Однак у 1979 році метод аналізу зчеплення був удосконалений і повторно проведені дослідження зчеплення з іншими маркерами не підтвердили цю інформацію [24].
Точно локалізувати ген ФАГ вдалося лише в 1984 році, коли стало можливим застосовувати технології рекомбінатних ДНК. Лідскі і співавтори гібридизирували по Саузерну ДНК-панелі клітинних гібридів людина/миша з отриманими на той час клонами кДНК гена ФАГ людини. Максимальний гібридизаційний сигнал був отриманий для клонотеки хромосоми 12. У результаті наступної гібридизації in situ із хромосомою 12, у якої були делетировані різні ділянки, ген ФАГ був точно картований в області 12q22-24.1 [2].
У 1985 році Ву і співавтори, використовуючи клоновану кДНК кролика в якості специфічної гібридизаційної проби, виділили і клонували кДНК печінки людини (клон phPAH47), на наступний рік тим же колективом авторів була визначена повна послідовність нуклеотидів кДНК, а ще через рік - і повну послідовність гена ФАГ [25, 26].
Бібліотека кДНК кліток печінки людини, що містить 14 мільйонів рекомбінантів була сконструйована в клонованому експрессируючому векторі ![]() gt11. Для одержання повної послідовності кДНК гена ФАГ авторами було проскриновано 100 000 рекомбінантів і отримано 100 позитивних гібрідизаційних сигналів. Чотири клони мали розмір в будованого фрагмента 2000 - 2500 п.н., що відповідає отриманому раніше методом гібридизації по Нозерну розміру мРНК фенілаланінгідроксилази. Ці 4 клони були оброблені рестриктазою Eco1, вбудований фрагмент був витягнутий і вбудований по Eco1 сайту плазміди pBR322. Використовуючи метод сиквенса, запропонований Максамом і Гилбертом, автори визначили розмір і послідовність нуклетидів кДНК гена ФАГ людини [25, 26]
gt11. Для одержання повної послідовності кДНК гена ФАГ авторами було проскриновано 100 000 рекомбінантів і отримано 100 позитивних гібрідизаційних сигналів. Чотири клони мали розмір в будованого фрагмента 2000 - 2500 п.н., що відповідає отриманому раніше методом гібридизації по Нозерну розміру мРНК фенілаланінгідроксилази. Ці 4 клони були оброблені рестриктазою Eco1, вбудований фрагмент був витягнутий і вбудований по Eco1 сайту плазміди pBR322. Використовуючи метод сиквенса, запропонований Максамом і Гилбертом, автори визначили розмір і послідовність нуклетидів кДНК гена ФАГ людини [25, 26]
Ген фенілаланінгідроксилази має розмір 90 т.п.н. До складу гена входить 13 екзонів і інтронів [25]. Середня довжина екзонів приблизно 114 п.н., самий короткий 9-ий екзон - 57 п.н., найбільш довгий 6-ий - 197 п.н. Тринадцятий екзон складається з 894-х нуклеотидів, але протеінкодируюча ділянка містить лише 42 нуклеотида. Розміри інтронів значно більше: мінімальна довжина інтрона 1 т.п.н. (сьомий інтрон), максимальна - 23.5 т.п.н. (третій інтрон). У 3'-частині гена зосереджені більш компактні інтрони, у 5'-частині - інтрони великих розмірів. Таким чином, сумарна довжина екзонних послідовностей складає 2.3 т.п.н., а інтронних 85 т.п.н. Показник співвідношення некодуючої частини гена до кодуючої для гена ФАГ - один з найбільших у порівнянні з іншими відомими на сьогодні генами еукаріот [25].
Було показано, що нуклеотидна послідовність гена фенілаланінгідроксилази людини мають 90%-у гомологію з аналогічним геном кролика [26] і 92,3%-у гомологію з геном ФАГ миші [27], більш того, деякі з описаних на сьогоднішній день мутацій гена ФАГ людини ідентифіковані й у миші [28].
Експресія гена ФАГ відбувається тільки в клітках печінки. Незначний рівень експресії відзначають вже в процесі ембріогенеза, починаючи з першого триместру вагітності, але система гідроксилірування ФА підключається до роботи тільки в постнатальний період [29].
1.4. Білковий продукт гена ФАГ
Білковим продуктом гена ФАГ є залізовмісний фермент - фенілаланінгідроксилази, що складається з 452-х амінокислотних залишків. Модель структури ферменту була створена в 1998-1999 р.р (Рис. 1.1) [30, 31].
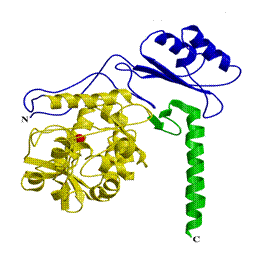

Рис. 1.1. Структура білка фенілаланінгідроксилази людини (модель). Червоний - ліганд заліза, жовтий - католітичний домен, фіолетовий - регуляторний домен, зелений – тетромеризующий домен [30, 31].
Субодиниці ферменту складаються з трьох доменів: регуляторний домен (амінокислоти 1-142), каталітичний домен (амінокислоти 143-406) і тетрамеризующий домен (амінокислоти 407-452) [29, 30, 31].
1.5. Поліморфні послідовності ДНК гена ФАГ
Під час вивчення молекулярної структури гена ФАГ було ідентифіковано кілька поліморфних ділянок ДНК, які можна розділити на наступні групи: 1) біалельні поліморфізми довжини рестрикційних фрагментів (ПДРФ); 2) мультиалельні поліморфізми, представлені в послідовності гена ФАГ мінісателлітним VNTR (варіююче число тандемних повторів) і мікросателлітним STR (короткі тандемні повтори) поліморфізмами; 3) однонуклеотидні поліморфізми (SNP).
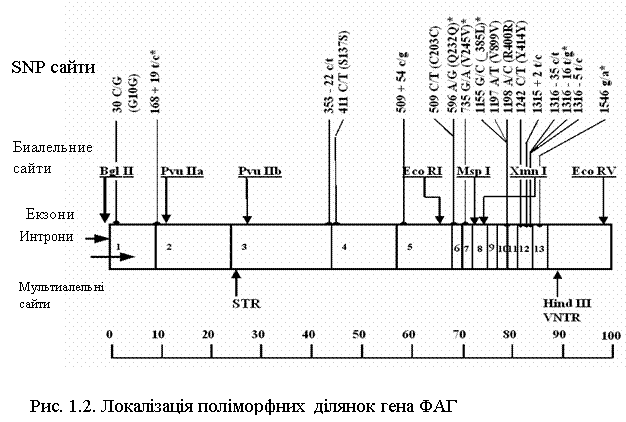
Історично першими були ідентифіковані ПДРФ поліморфізми гена ФАГ. У процесі дослідження кДНК було знайдено вісім поліморфних сайтів рестрикції для семи різних ендонуклеаз: Bgl, PvuII, EcoRI, Xmn, Msp, EcoRV, HindIII [32]. На малюнку 1.2 представлений весь спектр і локалізація поліморфізмів гена ФАГ [33]. Точна локалізація цих ділянок була проведена за допомогою методу блот-гібрідизації з кДНК-зондами, що являють собою різні ділянки гена.
Поліморфізми довжини рестрикційних фрагментів (ПДРФ) ФАГ локусу утворюють два кластери на 5'-кінці і 3'-кінці гена, відстань між якими приблизно 30 т.п.н. До складу 5'-кластера входять екзони з 1 по 3 з поліморфними сайтами для ендонуклеаза Bgl , PvuII (а), PvuII(б). Його розмір складає приблизно 40 т.п.н. 3'-кластер з поліморфними сайтами для ендонуклеаза EcoRI , Xmn, Msp, EcoRV, HindIII включає екзони з 6 по 13 і має розмір 30 т.п.н.[25]
При складанні рестрикційної карти гена ФАГ для HindIII поліморфізму, локалізованого в 3'-частині гена, було відзначене існування трьох алельних варіантів – явище не типове для біалельних ПДРФ поліморфізмів [32, 34]. Були визначені такі розміри рестрикційних фрагментів, як HindIII 3,4 т.п.н.; HindIII 3,6 т.п.н. і HindIII 3,8 т.п.н. [35]. Для подальшого вивчення цієї ділянки гена ФАГ ДНК-зонди, що містять 13-й екзон і 3 т.п.н. 3'-нетрансльованої послідовності проклонували в раніше сконструйованій плазміді pKSP23E12. Це дозволило більш точно локалізувати поліморфний регіон і обмежити його ділянкою розміром 0,6 т.п.н [36]. При секвенирувані цього фрагмента визначили його первинну структуру і показали наявність Ат-богатого (70%) мінісателіта на відстані 3 т.п.н від 13-го екзона в 3'-нетрансльованій ділянці. Розмір одиниці цього VNTR-поліморфізму, що повторювалася, складає 30 п.н. Кількість тандемних повторів варіірує від 3-х до 12-ти. Таким чином, отримані результати пояснюють наявність трьох аллелей HindIII поліморфізму існуванням мінісателітного VNTR поліморфізму в цьому регіоні, алель із трьома повторами якого відповідає HindIII 3,4; 5; 6, 7, 8, 9 повторів – HindIII 3,6 і 12 повторів – HindIII 3,8 [36]. VNTR-поліморфізм має високу інформативність [33, 37].

Для більшості мутацій гена ФАГ була показана асоціація з визначеними аллелями VNTR-поліморфізму, що свідчить про те що, які б події не призвели до виникнення мультиалельності в цьому локусі, вони передували процесу виникнення відомих на сьогоднішній день мутацій [36].
У транскрибіруємій послідовності гена ФАГ було ідентифіковано 40 SNP поліморфізмів. Ці поліморфізми являють собою одиничні нуклеотидні заміни, але не ПДРФ. Дев'ять SNP локалізовані в інтронних послідовностях і 31 - в екзонних [38]. Ці поліморфізми називають такими що мовчать, тому що заміна одного нуклеотиду в амінокислоті при SNP поліморфізмі не призводить до виникнення мутації, наприклад Q232Q.
1.6. Мутації гена фенілаланінгідроксилази.
Як відзначалося вище, причиною розвитку ФКУ є дефект синтезу ферменту ФАГ, а це, у свою чергу, є наслідком мутацій у гені ФАГ. До 2001 року були ідентифіковані більш 600 мутацій, переважна більшість яких відноситься до класу міссенс мутацій, тобто мутацій, що призводять до одиничної амінокислотної заміни. Крім міссенс мутацій є нонсенс мутації, що приводять до виникнення стоп кодона, а також делеції і мутації донорних і акцепторних сайтів сплайсингу [38].
Розповсюдження мутацій по місцю локалізації надана в таблиці 1.1 [38]. Частоти мутацій гена ФАГ у хворих ФКУ в Україні надана в таблиці 1.2 [33, 39]. У зв'язку з тим, що найбільше число мутацій знаходиться в сьомому екзоні, сьомий екзон вимагає особливої уваги. Наш інтерес до сьомого екзона підсилюється тим фактом, що за допомогою однієї ампліфікації і 6 рестрикцій ми можемо індифікувати 6 мутацій (R261Q, G272X, P281L, R261X, S273F, R252W).
Таблиця 1.1. Поширення мутацій по місцю локалізації| Локалізація | Кількість мутацій |
| 1 екзон | 7 |
| 3 екзон | 38 |
| 5 екзон | 1 |
| 7 екзон | 69 |
| 11 екзон | 39 |
| 12 екзон | 22 |
| 10 інтрон | 1 |
| 12 інтрон | 1 |
Таблиця 1.2. Частоти мутацій гена ФАГ у хворих на ФКУ в Україні
|
Мутація |
Частота % |
|
R408W |
57 |
|
R158Q |
3,7 |
|
R252W |
2,9 |
|
P281L |
2,3 |
|
Y414C |
1,6 |
|
Ivs10nt546 |
1,5 |
|
Ivs12nt1 |
1 |
|
R261Q |
1,2 |
|
G272X |
0,6 |
|
S237F |
0,6 |
|
R413P |
0,5 |
|
Не ідентифіковані |
27 |
Найбольший інтерес в Україні й у багатьох країнах Європи має мажорна мутація R408W [33,40]. Також становлять інтерес такі розповсюджені мутації як: Ivs12nt1, R158Q, Y414C, Ivs10nt546. Існує багато методів аналізу цих мутацій такі як: ПЛР, ИФА і блот гібридизація. Найменша собівартість одного аналізу, чутливість і точність досягається за допомогою полімеразної ланцюгової реакції, тому ми віддамо перевагу саме ПЛР.
1.7. Полімеразна ланцюгова реакція
Полімеразна ланцюгова реакція – це метод, що імітує природну реплікацію ДНК і дозволяє знайти єдину специфічну молекулу ДНК у присутності мільйонів інших молекул.
Відкриття методу полімеразної ланцюгової реакції (ПЛР) стало одним з найбільш видатних подій в області молекулярної біології за останні десятиліття. Це дозволило підняти медичну діагностику на якісно новий рівень.
Основні принципи використання праймерів (коротких штучно синтезованих молекул ДНК) і склад інгредієнтів, що входять у реакційну суміш для одержання копій ДНК, уперше були описані Kleppe із соавторами у 1971 році. Однак тоді ще не була продемонстрована основна риса ПЛР - експонентне збільшення кількості копій фрагмента вихідної ДНК.
У 1983 році співробітник фірми «Cetus» Kary Mullis запропонував метод, що став надалі відомим як полімеразна ланцюгова реакція. Суть методу полягає в багаторазовому копіюванні (ампліфікації) у пробірці визначених ділянок ДНК у процесі температурних циклів, що повторюються. На кожнім циклі ампліфікації синтезовані раніше фрагменти знову копіюються ДНК-полімеразою. Завдяки цьому відбувається багаторазове збільшення кількості специфічних фрагментів ДНК, що значно спрощує подальший аналіз.
Слід зазначити, що це відкриття супроводжувалося розвитком технологій. Зокрема, з'явилися прилади, що дозволяли автоматично синтезувати одноланцюгові фрагменти ДНК (олігонуклеотиди). У той же період були виявлені унікальні мікроорганізми, що живуть у гейзерах. Їхня ферментативна система, зокрема ДНК-полімераза, витримує високі температури гарячих джерел і зберігає свою біологічну активність аж до 95°С, що є необхідною умовою для проведення полімеразної ланцюгової реакції [41].
Результатом відкриття ПЛР стало майже негайне практичне застосування методу. У 1985 році Saiki із соавторами опублікували статтю, у якій була описана ампліфікація геномної послідовності ![]() -глобина [42]. З цього моменту кількість публікацій, у яких автори повідомляли про застосування ПЛР у своїх роботах, стало збільшуватися в геометричній прогресії. Метод придбав таку популярність, що сьогодні уже важко представити роботу в області молекулярної біології без його використання. Особливо бурхливий розвиток метод полімеразної ланцюгової реакції одержав завдяки міжнародній програмі «Геном людини». Були створені сучасні лазерні технології сиквенування (розшифровки нуклеотидних послідовностей ДНК). Якщо в недавньому минулому для розшифровки послідовності ДНК розміром у 250 пар нуклеотидів (п.н.) був потрібен тиждень, то сучасні лазерні сиквенатори дозволяють визначати до 5000 п.н. у день. Це у свою чергу сприяє значному росту інформаційних баз даних, що містять послідовності ДНК різних біологічних об'єктів. В даний час запропоновані різні модифікації ПЛР, показана можливість створення тест-систем для виявлення мікроорганізмів, виявлення крапкових мутацій, описані десятки різних застосувань методу.
-глобина [42]. З цього моменту кількість публікацій, у яких автори повідомляли про застосування ПЛР у своїх роботах, стало збільшуватися в геометричній прогресії. Метод придбав таку популярність, що сьогодні уже важко представити роботу в області молекулярної біології без його використання. Особливо бурхливий розвиток метод полімеразної ланцюгової реакції одержав завдяки міжнародній програмі «Геном людини». Були створені сучасні лазерні технології сиквенування (розшифровки нуклеотидних послідовностей ДНК). Якщо в недавньому минулому для розшифровки послідовності ДНК розміром у 250 пар нуклеотидів (п.н.) був потрібен тиждень, то сучасні лазерні сиквенатори дозволяють визначати до 5000 п.н. у день. Це у свою чергу сприяє значному росту інформаційних баз даних, що містять послідовності ДНК різних біологічних об'єктів. В даний час запропоновані різні модифікації ПЛР, показана можливість створення тест-систем для виявлення мікроорганізмів, виявлення крапкових мутацій, описані десятки різних застосувань методу.
Аналіз виявлення крапкових мутацій на базі полімеразної ланцюгової реакції складається з наступних етапів:
- виділення ДНК матриці;
- ампліфікація;
- детекція ампліфікованих продуктів;
- повний або частковий аналіз нуклеотидної послідовності продуктів ПЛР такими методами як: рестрикційний аналіз, гетеродуплексний аналіз, електрофоретичне фракціонування.
1.7.1. Компоненти ампліфікаційної реакційної суміші
Для проведення полімеразної ланцюгової реакції необхідна наявність у реакційній суміші ряду компонентів:
- Праймери;
- Taq-полімераза;
- dNTP;
- Буфер;
- Mg2+;
- Аналізований зразок;
- ПЛР добавка.
Праймери для ампліфікації in vitro.
Праймери – штучно синтезовані олігонуклеотиди, що мають, як правило, розмір від 15 до 30 п.н., ідентичні відповідним ділянкам ДНК-мішені. Їх можна синтезувати за допомогою автоматичних синтезаторів ДНК. Часто в 5'-кінцеву ділянку праймерів для спрощення клонування ПЛР-ампліфікованої ДНК уводять сайти розпізнання для рестриктирующих ендо-нуклеаз. У 3'-кінцеву ділянку праймерів не можуть бути внесені ніякі заміни, тому що при низькій концентрації нуклеозидтрифосфатів таке порушення блокує ампліфікацію. Якщо концентрація нуклеозидтрифосфатів знаходиться на рівні, звичайному для ПЛР, то невелике порушення гомології між 3'-кінцем праймера і матрицею не позначається на ході реакції. Таким чином, за допомогою ПЛР можна розрізнити аллелі, що відрізняються всього одним нуклеотидом.
Підбір праймерів - ключова ланка ПЛР, оскільки саме ними визначається можливість ампліфікації і виявлення потрібної послідовності, а також надзвичайна гнучкість методу. Просте варіювання праймерів дозволяє виявляти багато патогенних мікроорганізмів і генетичних порушень при мінімальних змінах у методиці [41].
Необхідно відзначити, що праймери можуть відпалюватися один з другим, утворюючи праймери-димери, і можуть самі на себе, утворюючи петлі [43]. І те, і інше призводить до значної витрати праймерів на синтез побічних (неспецифічних) продуктів реакції і, як наслідок, значно зменшення чутливості системи. Це або ускладнює, або унеможливлює проведення аналізу [44].
Процес спарювання праймерів розглянутий у статтях [46, 47], де розглянута термодинаміка утворення петель.
Довжина ПЛР-продукту визначається сумою розміру праймерів і відстанью між їх 3'-кінцями й у більшості випадків лежить у діапазоні 100 - 300 нуклеотидів. Швидкість реплікації за допомогою ДНК-полімерази Tag складає 35 - 100 нуклеотидів на секунду [47].
Оцінка кількості праймера може бути визначена за наступними міркуваннями. Якщо в 100µl буде синтезовано 3µg 500bp продукти, то на це повинно піти праймера: 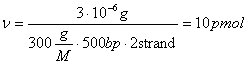 . Використовується діапазон концентрацій від 0.1 до 0.6 µM.
. Використовується діапазон концентрацій від 0.1 до 0.6 µM.
Краще, якщо пара "збалансована", тобто різниця температур плавління, не перевищує 2-4ºС.
При створенні ПЛР-тест-системи однією з основних задач є правильний підбір праймерів, що повинні відповідати ряду критеріїв:
1. Праймери повинні бути специфічні. Особливу увагу приділяють 3'-кінцям праймерів, тому що саме з них Taq-полімераза починає добудовувати комплементарний ланцюг ДНК. Якщо їхня специфічність недостатня, то, ймовірний синтез неспецифічної ДНК (коротких чи довгих фрагментів). На електрофорезі вони спостерігаються у вигляді важких або легких додаткових смуг, іноді шмерів, що виглядають суцільним мазком. Це заважає оцінці результатів реакції, тому що легко переплутати специфічний продукт ампліфікації з неспецифічним. Частина праймерів і ДНТФ витрачається на синтез неспецифічної ДНК, що призводить до значної втрати чутливості.
2. Праймери не повинні утворювати димери і петлі, тобто не повинно утворюватися стійких подвійних ланцюгів у результаті відпалу праймерів самих на себе або один з другим.
3. Область відпалу праймерів повинна знаходитися поза зонами мутацій, делецій чи інсерцій. При влученні на таку зону віджиг праймерів відбуватися не буде, і, як наслідок, буде отриманий ложнонегативний результат.
Праймери відіграють ключову роль в утворенні продуктів реакції ампліфікації. Правильно підібрані праймери забезпечують специфічність і чутливість тест-системи.
Фермент для ампліфікації in vitro
У 1980 році полімеразна активність була відкрита в деяких формах термофільних бактерій. Таg-полімераза була виділена з бактерій виду Thermus aquaticus, здатних зростати при температурі 70-75°С. Молекулярна вага очищеного протеїну 94 кД і оптимальна температура полімеразної активності 70 - 80°С. При 90°С активність ферменту зменшується, але полімераза не денатурується, при зниженні температури до 70-80°С рівень активності відновлюється.
Tag-полімераза має дуже високу швидкість синтезу. При оптимальних умовах фермент може добудовувати до 150 основ на секунду. При низькій температурі активність падає до 2 основ на секунду. Час напівжиття Таg-полімерази при 95°С складає 40 хвилин [44].
Для проведення ПЛР-тестів звичайно використовують ДНК-полімеразу, виділену з Thermus aquaticus. У цього ферменту відсутня коригувальна 3'-4'-5'-активність і частота помилок при зчитуванні досягає 1/10 000 нуклеотидів. Цієї точності (хоча вона і нижче, ніж у багатьох інших полімераз) досить для того, щоб виникаючі при реплікації послідовності-мішені, помилки не створювали особливих проблем.
Оптимальна кількість ферменту залежить від кількості матриць і довжини ПЛР продукту.
1U – це та кількість ферменту, яке зв'язує 10 нмоль дезоксінуклеотидтрифосфатів за 30 хв. при 74ºС [48].
Якщо використовується занадто маленька кількість Taq, вихід продукту знижується, причому це зниження тим серйозніше, чим більше розмір продукту. Не варто використовувати і занадто великий надлишок Taq полімерази, тому що при цьому спочатку з'являються високо- і низькомолекулярні шмери (перевищення ~ 2-4 рази), а при подальшому збільшенні концентрації ференту (перевищення ~ 4-16 разів) весь продукт стає надзвичайно маленьким.
Для рутинного використання часто використовують суміш Taq:Pfu=100:1, у якої точність синтезу практично така ж, як у Pfu полімерази. У зручності використання така суміш не поступається Taq полімеразі.
Так як молярна вага Ampli Taq "Perkin-Elmer" дорівнює 94kDa та 1mg - 250000u, то 1u відповідає 4ng, або 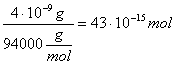 =43fmol.
=43fmol.
Taq полімераза володіє 5'-3' екзонуклеазною активністю. Ця активність може викликати проблеми, пов'язані з деградацією 5' кінців продукту.
Точність Taq полімерази залежить від концентрації Mg2+, dNTP's, збалансованості dNTP's та pH. У середньому частота помилок нижче, ніж 1 заміна на 100-300bp.
Так як Taq полімераза з високою частотою включає неправильні нуклеотиди, то для правильного визначення послідовності потрібно сиквенувати не одну клонованау матрицю, а кілька. Якщо ПЛР-продукти сиквенувати безпосередньо, то послідовність виходить точною, тому що для того, щоб знайти мутацію, вона повинна бути присутнім у >10% матриць. Це можливо лише в тому випадку, якщо мутація відбулася в першому циклі, а матриць при цьому було не більш 5 штук.
Для Phusion Polymerase оптимальна кількість полімерази, як правило, лежить у межах 0,5 – 2 U на 50 мкл [48].
Суміш дезоксінуклеотидтрифосфатів
Суміш дезоксінуклеотидтрифосфатів (дНТФ) – дезоксіаденозинтри-фосфат (дАТФ), дезоксігуанозинтрифосфат (дГТФ), дезоксіцитозинтрифосфат (дЦТФ) і дезоксітимидинтрифосфат (дТТФ) – «будівельний матеріал», який використовуеться полімеразою для синтезу другого ланцюга ДНК. Важливо підтримувати концентрацію значно вище, ніж Km відповідної полімерази. Чим менше концентрація dNTPs, тим вище точність синтезу. Діапазон використовуваних концентрацій складає 50-500 µM. Високі концентрації дНТФ можуть привести до включення не комплементарних нуклеотидів у наслідку термодинамічної байдужності який нуклеотид включити у послідовність, що добудовуєься [49]. Суміш дезоксінуклеотидтрифосфатів володіють значною буферною місткістю, що зменшується з кожним циклом ампліфікації [44]. Для Phusion™ DNA Polymerase рекомендується використовувати таку кількість дезоксінуклеотидтрифосфатів, щоб їхня концентрація в ампліфікаційній суміші становила 20 мкм [48].
Буфер для ампліфікації
Буфер - суміш катіонів і аніонів у певній концентрації, що забезпечує оптимальні умови для реакції, а також стабільне значення рН середовища [42]. Багато полімераз мають свої буфери. Буфери розрізняються по призначенню. Компанія Finnztmes рекомендує використовувати різні буфери в залежності від співвідношення AT до GC. Тому що рівень помилок полімерази з GC буфером (9,5.10-7) набагато вище, ніж полімерази з HF буфером (4,4.10-7), GC буфер рекомендується використовувати, коли HF не працює [48].
Концентрація іонів магнію.
Так як ДНК полімераза магній - залежний фермент, то концентрація іонів Mg2+ є важливим параметром ампліфікації [48]. На молекулярному рівні: Mg2+ утворює комплекси з dNTP (саме ці комплекси є субстратом для Taq pol). C Mg2+ стехіометрично зв'язуються dNTP's, EDTA, (PO4)3-. Так як підвищення концентрації Mg2+ викликає підвищення температури плавлення ДНК [44, 50], підвищення концентрації Mg2+ призводить до стабілізації комплексів ДНК-праймерів з неповною гомологією, що знижує специфічність ампліфікації [48]. Надлишок іонів стабілізує двухланцюгову ДНК і запобігає повній денатурації ДНК. Процес денатурації ДНК розглянут у роботі [51], а також у розділі 1.7.2. Оптимальна концентрація іонів Mg2+ залежить від концентрації дезоксинуклеотидтрифосфатів, ДНК-матриці і складу буфера. У загальному випадку для стандартної ПЛР оптимальна концентрація іонів Mg2+ лежить у межах від 0,5 до 1 мМ більше спільної концентрації дезоксінуклеотидтрифосфатів. Якщо праймери і (або) ДНК утримують хелати, такі як EDTA чи EGTA, концентрація Mg2+ повинна бути збільшена. У разі потреби подальшої оптимізації умов, рекомендується збільшувати концентрацію Mg2+ із кроком 0,2 мкМ. [48]. Занадто низька концентрація Mg2+ обумовлює низький вихід продукту, у той час як занадто висока концентрація Mg2+ приводить до зниження виходу ПЛР-продукту. На малюнку № 1.3. представлений вплив концентрації іонів Mg2+ на ампліфікацію. Для даної системи оптимальна концентрація іонів Mg2+ складає 2,5 млмоль/л [50]. Також необхідно відзначити, що висока концентрація Mg2+ може призвести до неспецифічної ампліфікації [44].
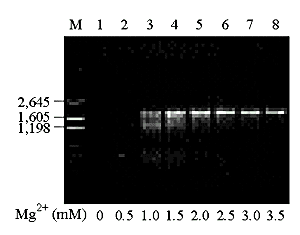
Рис. 1.3. Вплив концентрації іонів Mg2+ на ампліфікацію.
ДНК матриця.
Матриця це зразок, підготовлений до внесення в реакційну суміш, що може містити цільову ДНК, що служить мішенню для наступного багаторазового копіювання. При відсутності ДНК-мішені специфічний продукт ампліфікації не утвориться.
В якості матриці може виступати:
1. Геномна ДНК ссавців < 0.5-1µg;
2. Геномна ДНК бактерій ~1-10ng;
3. ДНК фага лямбда. Досить 1µl з 200µl елюції одиночної бляшки;
4. ПЛР продукт розведений у ~ 106 разів.
5. Плазмідна ДНК:
5.1. 0.1-1.0µl нічної культури,
5.2. ~50 нг очищеної суперскручений ДНК (така велика кількість береться через те, що pДНК - кільцева і при зниженні температури вона ренатурирует, не залишаючи праймеру шансів зв'язатися з нею. Якщо хочеться зменшити кількість внесеної pДНК, потрібно проводити попередню лужну денатурацію. Для цього необхідно взяти 4µl pДНК і змішати з 1µl 1N NaOH, отриманий розчин витримати при 65-85ºС протягом 5 хвилин і різко охолоджують до 0ºС (чи заморозити) і додати 1µl 1N соляної кислоти.
ПЛР добавка
ПЛР добавки - речовини, що поліпшують специфічність (і/або) вихід ПЛР. Для більшості ампліфікацій ПЛР добавки не використовуються. При ампліфікації GC-багатих ділянок, неякісних зразків ДНК і ампліфікації фрагментів більше 1 т. п. н. рекомендується використовувати допоміжні речовини, що приведені в таблиці 1.4 та 1.5 [44].
Важливо відзначити, що використання цих речовин в оптимизированной реакції може викликати погіршення виходу. Це пояснюється тим, що - зазначені речовини змінюють взаємодію між праймерами і матрицею і між матрицею і полімеразою.
У таблиці № 1.6. наведенні речовини, що можуть бути присутні в ампліфікаційній системі та їх дію на систему.
Таблиця 1.4. Допоміжні речовини, що використовуються для ампліфікації
|
Речовина: |
Використовувані концентрації: |
Механізм дії: |
|
Betain Na |
0.5-2M |
стабілізація ферменту, зниження Tm ДНК, вирівнювання Tm AT- и GC-багатих послідовностей |
|
BSA |
~0.1 µg/ml |
стабілізація ферменту |
|
DMSO |
Згідно [44] (оптим. ~5%). Згідно [48] (оптим. ~3%) |
Допомагає денатурувати CG-богатую ДНК матрицю. 10% DMSO знижує температуру плавлення на 5,5-6,0ºС [52] |
|
Formamid |
1-5% |
Допомагає денатурувати CG-богатую ДНК матрицю. |
|
Glycerol |
5-20% |
стабілізація ферменту |
|
NP-40, Tween 20, Triton X-100 |
0.1-0.5% |
стабілізація ферменту |
|
Tetramethylammonium chloride (TMA chloride) |
0.01-0.1mM |
підвищення специфічності |
|
ssДНК binding protein E.coli |
5 µg/ml |
стабілізація ssДНК |
|
ssДНК binding protein T4 gene 32 protein |
поліпшує вихід в концентраціях 0.5-1.0mM. |
стабілізація ssДНК |
Таблиця 1.5. Допоміжні речовини, що використовуються для ампліфікації
|
Речовина |
Максимальна |
90% інгибування |
|
|
ефективність |
специфічність |
||
|
TMA chloride |
1.9 (5 mM) |
0.5 (20 mM) |
(35 mM) |
|
TMA oxalate |
2.2 (2 mM) |
1.0 (2 mM) |
(9 mM) |
|
TMA acetate |
1.0 (<10 mM) |
0.4 (20 mM) |
(40 mM) |
|
TMA hydrogen sulfate |
1.2 (0.5 mM) |
1.0 (50 mM) |
(70 mM) |
|
Ammonium chloride |
1.0 (<20 mM) |
0.2 (<20 mM) |
(50 mM) |
|
Benzyldimethyl-hexadecylammonium chloride |
1.1 (0.2 mM) |
0.4 (1 mM) |
(1.0 mM) |
|
HTA bromide |
1.2 (0.05 mM) |
0.5 (0.5 mM) |
(0.8 mM) |
|
HTA oxalate |
1.0 (<0.2 mM) |
0.28 (<0.2 mM) |
(0.5 mM) |
|
Betaine monohydrate |
1.1 (100 mM) |
0.4 (750 mM) |
(900 mM) |
|
Dimethyl sulfoxide |
1.0 (<1.4 M) |
0.6 (1.4 M) |
(1.6 M) |
|
Formamide |
1.4 (0.5 M) |
0.8 (1 M) |
(2.0 M) |
|
ПЛР без добавок |
1.0 |
0.2 |
|
У дужках наведенна концентрація речовин, що призводить до зазначеного ефекту. Число перед дужками означає відносну ефективність дії. Специфічність оцінюється як частка ДНК, що приходиться на специфічну смугу. TMA похідні практично не змінюють оптимальну температуру відпалювання [53].
Таблиця 1.6. Вплив речовинин які можуть потрапити у ампліфікаційну суміш.
| Речовина | Дія |
| Агароза | не заважає до ~1%. |
| АА акриламід лінійний | не заважає (в низької концентрації). |
| AcONa (pH 5.0) | починає інгибувати при > 5mM. |
| BSA ацетильований | 10µg/ml – інгібує |
| BSA heat інактивований | 10µg/ml - не заважає |
| Vanadil ribonucleoprotein complexes | не заважають при < 0.04mМ, інгибує при > 0.4mМ; |
| Гемоглобін (концентрація в крові ~160mg/ml) | зменшує вихід при >1mg/ml |
| Гепарин | інгібує ПЛР при концентрації > 0.15U/ml гепарин використовується для запобігання згортання крові 14.3 u/ml, не усувається органічними розчинниками і переосадженням. |
| EDTA | зв'язується з Mg2+, та починає інгибувати при концентрації >0.5mМ, при концентрації 1mМ ПЛР не здійснюється. |
| DEPC | інгібує реакцію |
| Желатин | 10µg/ml - не заважає |
| Ізопропанол | інгибування при концентраціях >1% (більш сильний інгібітор, чим етанол) |
| Олія | може усувати інгибуючий ефект деяких забруднень |
| NaCl | помітно інгібує при концентрації 25mМ |
| RT суміш | не впливає на ПЛР при концентрації 10%, починає інгибувати при концентрації >15% |
| RNA | інгибує при концентрації > 0.5 мкг на 20µl; інгибуючий ефект можна усунути додаванням у реакцію RNase |
| Сахароза | не заважає до 30%. |
| Фенол | зменшує вихід при концентрації >0.2%, ПЛР не здійснюється при концентрації 0.5% |
| Ficoll 400 | не заважає до концентрації 1% (більша концентрація інгібує ПЛР) |
| Етанол | для деяких реакцій - стимулюючий ефект при 1%, для інших - інгибування при концентраціях >1% |
| Хлороформ | не впливає на ПЛР реакцію навіть при концентрації 5%. |
| Цитрат Na | не впливає при 1mМ |
| Меланін | помітно впливає від 0.01µg/µl, інгібує: 0.08µg/µl |
| Polyvinilpyrollidon (PVP) | не інгібує принаймні до 2% |
1.7.2. Температурний режим ампліфікації.
В процесі реакції ампліфікації з ДНК відбувається ряд подій, що забезпечуються певними температурними циклами. Кожний цикл ампліфікації складається з трьох етапів: денатурація, віджиг праймерів, елонгація, що будуть розглянуті далі. Як правило використовують 2-5 хвилинне попереднє нагрівання.
1. Денатурація. Попереднє нагрівання здійснюють для прогріву проби та денатурації двухланцюгової ДНК. Температуру і час попереднього прогріву вибирають виходячи зі складу ДНК-матриці і властивостей полімерази. Для Tag полімерази рекомендується здійснювати попередній прогрів до 94ºС протягом 30 - 300с. Експозиція 300 с використовується для ампліфікації CG-багатих ділянок. Однак, якщо частка CG-основ у ПЛР продукті перевищує 75%, то рекомендується використовувати прогрів до 98ºС. Також як усі молекули ДНК не плавляться одночасно, так і кожна окрема молекула вся не плавиться [54]. Молекула ДНК плавиться поступово Рис. 1.4, і в першу чергу в ділянках з високою часткою AT основ з утворенням вільних кінців і внутрішніх циклів, що представлені на малюнку № 1.5. Імовірність утворення внутрішнього циклу (рис. 1.6.) розглянута в роботі [55].
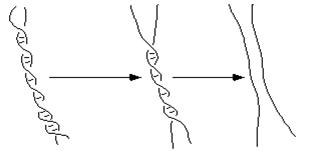
Рис. 1.4. Процес денатурації подвійної спіралі ДНК.
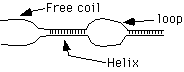
Рис. 1.5. Утворення вільних кінців, спіралі і внутрішніх циклів при денатурації подвійної спіралі ДНК.
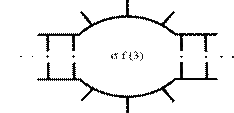

Рис. 1.6. Утворення внутрішнього циклу.
Однак використання високої температури має ряд недоліків. Один з основних недоліків - це зниження часу напіврозпаду полімерази. Час напіврозпаду Taq полімерази в залежності від температури представлений в таблиці № 1.7. Залежність часу напіврозпаду Taq полімерази від буфера при 96.5ºС представлена в таблиці № 1.8.
Так при 98ºC час напіврозпаду Tag Pol складає 5 хв, що обумовлює не можливість її використання. У таких випадках рекомендується використовувати Gold ДНК Polymerase або Phusion ДНК Polymerase. При використанні Phusion ДНК рекомендується проводити 30-180 секундний попередній прогрів при температурі 98ºС. Якщо проводити масовий скринінг CG багатої області, то використання Gold ДНК Polymerase або Phusion ДНК Polymerase може значно підвищити собівартість дослідження. Альтернативним варіантом може бути використання суміші Gold ДНК Polymerase або Phusion ДНК Polymerase з Tag-полімеразою при цьому необхідно використовувати денатуруючі агенти такі як бетаін, DMSO або інші. Використання таких речовин дозволяє знизити температуру денатурації CG-багатої ділянки до 94ºС.
Таблиця 1.7. Час напіврозпаду Taq полімерази в залежності від температури.
|
Температура ºС |
Час хв. |
|
92.5 |
130 |
|
95.0 |
40 |
|
97.5 |
5-6 |
Таблиці № 1.8. Швидкість руйнування Taq полімерази в різних буферах при 96.5ºС.
|
Буфер |
Час напіврозпаду хв. |
|
H2O |
2.5 |
|
TE |
7.5 |
|
20mM Tris, pH9.0 |
10 |
Якщо матриця одноланцюгова або ПЛР-продукт, то попередній прогрів можна не проводити.
Так як кільцева pДНК уміє ренатурувати після теплової денатурації, то рекомендується здійснювати попереднє нагрівання при 95ºС протягом 1 хв.
2. Відпалювання праймерів. На другому етапі праймери приєднуються до одноланцюгової ДНК-мішені. Цей процес має назву «відпалювання» (від англ. «annealing»).
Оптимальна температура і час відпалювання праймерів забезпечують ефективну ПЛР. Якщо температура відпалювання праймерів буде занижена, то це може призвести до неспецефічної ампліфікації, а якщо завищена до зниження виходу ПЛР-продукту. На малюнку № 1.7. наведена електрофореграмма з ПЛР продуктами, отриманими при ампліфікації п'яти різних проб ДНК. Перші п'ять проб були отримані при температурі відпалювання 610С, наступні п'ять проб при температурі 590С [57]. На даному малюнку видно як збільшення температури призводить до зменшення виходу ПЛР-продукту, це може бути пов'язано з неповною гомологією праймерів, низькою якістю ДНК-матрици та іншими факторами.
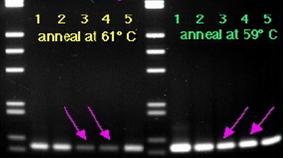
Рис.1.7. Залежність виходу ПЛР-продукту від температури відпалювання праймерів.
На малюнку 1.8. зображно варіювання температури плавлення, кількість ампліфікованих локусів і концентрації буфера, які змінюються в рівнобіжних реакціях. Доріжки 1-9 кожного гелю демонструють ПЛР в однократному буфері. Доріжки 10-12 кожного гелю демонструють ПЛР у дворазовому буфері. На не позначених доріжках нанесений маркер який містить ПЛР-продукт довжиною 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 т. п. н. П'ять стрілок ліворуч від першого гелю показують очікуваний ПЛР-продукт суміші С (п'ять продуктів). Самий довгий специфічний продукт на кожнім гелі маркірований червоною стрілкою. Рожеві стрілки вказують на інтенсивний, важкий не специфічний ПЛР продукт. Жовті стрілки вказують на два додаткових продукти обумовлені сумішшю праймерів С. Сині стрілки вказують на продукт Y. Ми можемо спостерігати багато неспецифічних смуг при температурі відпалювання 48 °C, що зникають при збільшенні температури відпалювання. Хоча праймери фланкуючі ПЛР продукт Y знаходяться у всіх сумішах в однаковій концентрації, найбільш стійкий ПЛР продукт спостерігається в реакції ампліфікації з однією парою праймерів. Потім йде мультиплексна ПЛР із п'ятьма парами праймерів. Найбільш слабкий продукт Y спостерігається в мультиплексній ПЛР із сімома парами праймерів. Дворазове збільшення концентрації ПЛР буфера дозволяє збалансувати вихід легких продуктів але в тойже час призводить до збільшення виходу важких не специфічних продуктів.
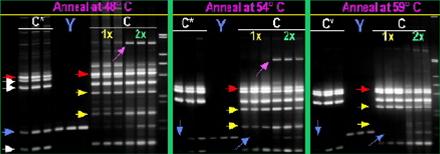
Рис. 1.8. Мультиплексна ампліфікація: суміші пар праймерів С*, “Y” та C на трьох різних ДНК матрицьях, з використанням трьох програм які відрізняються температурою відпалювання (48°C, 54°C, 59°C).
Відпалювання відбувається відповідно до правила компліментарності Чаргаффа, що означає, що в двухланцюговій молекулі ДНК навпроти аденина завжди знаходиться тимин, а напроти гуаніну – цитозин (рис. 1.9). Якщо ця умова не дотримана, то відпалювання праймерів не відбувається.
Рис. № 1.9. Принцип компліментарності й антипаралельності ланцюгів ДНК.
Визначення температури відпалювання праймерів.
У загальному випадку більш висока температура відпалювання обумовлює більш високу специфічність. Але як тільки температура відпалювання перевищує деяку критичну (для даної пари праймерів), кількість продукту починає різко знижуватися.
Температура оптимального відпалювання праймерів залежить від:
- полімерази;
- складу праймера (відношення CG до AT);
- послідовності праймера (nearest-neighbor взаємодії);
- концентрації Na+ [61];
- концентрації праймера;
- концентрації Mg2+
- присутності денатуруючих речовин таких як формамид, DMSO та іньші [58];
- послідовності між праймерами;
- pH;
- іонної сили ;
- концентрації ДНК;
- наявність інших праймерів;
- наявність інгібіторів ПЛР;
Перераховані вище фактори врахувати проблематично, тому температуру відпалювання праймерів визначають емпірично. При цьому як орієнтир використовують температуру плавлення праймера. При використанні Tag-полімерази рекомендують використовувати температуру відпалювання праймерів, розраховану за формулою (1.1). Фірма GENOSYS рекомендує використовувати температуру відпалювання на 5-100С нижче температури плавлення[58]. Для Phusion™ Polymerase рекомендується використовувати температуру відпалювання на 30С більше температури плавлення при довжині праймера більш 20 п.н. Це пояснюється тим, що Phusion™ Polymerase має здібність стабілізувати комплекс праймер-ДНК [48].
Tотжига = Tплавления - 50С (1.1)
Температура плавлення - це температура, при якій половина нуклеінових кислот знаходиться в одноланцюговому стані, а друга половина - у двухланцюговому.
Визначення температури денатурації комплексу ДНК-праймер.
Для коротких ДНК олігонуклеотидів (14 – 20 пар основ), при концентрації NaCl 0.9M рекомендується рівняння (1.2).
Tплавления =2(#А+#Т)+4(#G+#С) (1.2)
де: #А, #Т, #G, #С – колькість соответствующих нуклеотидів в последовательности праймера.
Для олігонуклеотидів довжиною більш 50 пар основ при ![]() рекомендується рівняння (1.3).
рекомендується рівняння (1.3).
![]() (1.3)
(1.3)
де: ![]() - молярна концентрація моновалентного катіона (у даному випадку
- молярна концентрація моновалентного катіона (у даному випадку ![]() ),
), ![]() і
і ![]() - частка залишків G і C – в олігонуклеотиді, L – довжина короткого ланцюга в дуплексі, F – відсоток формаміда в розчині. Дане рівняння не застосовується при
- частка залишків G і C – в олігонуклеотиді, L – довжина короткого ланцюга в дуплексі, F – відсоток формаміда в розчині. Дане рівняння не застосовується при ![]() [79].
[79].
Однак емпірично отримані рівняння (1.2) – (1.3) не адекватні даними, наведеним в таблиці № 1.9.
Таблиця 1.9. Темтература плавлення олігонуклеотиду в залежності від його послідовності [58].
|
№ проби |
Послідовність(5'-3') |
|
M |
Дов-жина |
GC |
|
1 |
AAAAACCCCCGGGGGTTTTT |
69.7 |
6103 |
20 |
50% |
|
2 |
ACGTACGTACGTACGTACGT |
57.2 |
6103 |
20 |
50% |
|
3 |
GATCGATCGATCGATCGATC |
64.5 |
6103 |
20 |
50% |
|
4 |
ATATATATATCGCGCGCGCG |
66.4 |
6103 |
20 |
50% |
Дані в таблиці № 1.9. вказують на те, що температура плавлення олігонуклеотиду залежить не тільки від його довжини і співвідношення AT до CG, але і від розташування нуклеотидів у ланцюзі. Це зв'язано з тим, що нуклеотиди своїми полярними і неполярними групами змінюють простір навколо себе і, таким чином, діють на сусідні нуклеотиди і на їхні водневі зв'язки. Судячи з таблиці № 1.9. ця дія достатньо велика. Для обліку впливу послідовності нуклеотидів на стійкість ДНК розроблена модель, що припускє, що на кожн нуклеотид вплива два його сусіди (Nearest Neighbor Model). Вплив кожного нуклеотиду на стійкість двухланцюгової ДНК оцінюється по енергії Гібса, а точніше по зміні енергії Гібса в реакції плавлення двухланцюгової ДНК. Негативне значення енергії Гібса є рушійною силою всіх процесів відомих науці. Якщо енергія Гібса дорівнює нулю, то система знаходиться в стані рівноваги. Якщо енергія Гібса позитивна, то протікає зворотний процес. Константа рівноваги є відношенням швидкості прямого процесу (реакції) до швидкості зворотного процесу (реакції), і може бути отримана з виразу (1.4).
![]() (1.4)
(1.4)
де: ![]() – зміна енергій Гібса в ході реакції, T – температура, R – універсальна газова постійна [51, 55, 66, 68].
– зміна енергій Гібса в ході реакції, T – температура, R – універсальна газова постійна [51, 55, 66, 68].
Необхідно відзначити, що, якщо концентрація одного з ланцюгів ДНК на багато переважае концентрацію комплементарної матриці (як у випадку присутніст праймера), то константа рівноваги бу дорівнювати рівноважному співвідношенню одноланцюгов молекул ДНК до дволанцюгових. На малюнку № 1.10. представлена практично отримана залежність частки двухланцюгових ДНК від температури.
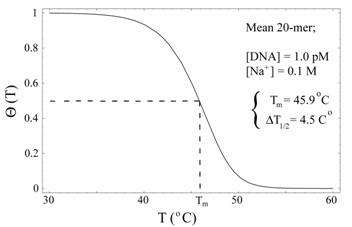
Рис. 1.10. Залежність частки двухланцюгової ДНК до всіх молекул ДНК в залежності від температури [55].
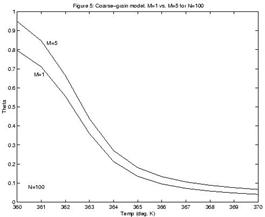
Рис. 1.11. Залежність частки двухланцюгової ДНК (Theta) від температури з різною часткою АТ основ. Розрахунок за допомогою апроксимации [54].
Різниця енергій Гібса може бути визначена з рівняння (1.5).
![]() (1.5)
(1.5)
де: ΔH – зміна ентальпії в ході реакції,
ΔS - зміна ентропії в ході реакції [51, 55, 59].
Зміна ентальпії й ентропії в ході реакції утворення комплексу ДНК-праймер може бути визначена з принципу адитивності, згідно якого ![]() ,
, ![]() [51]. Необхідно відзначити, що принцип адитивності може бути застосований для разрахунку зміни стійкості комплексу праймер одноланцюгова ДНК у біля температури плавлення, але він вимагає корекції ентальпії й ентропії. Необхідність корекції ентальпії й ентропії пов'язана з тим, що ентальпія й ентропія залежать від температури рівняння (1.6) і (1.7).
[51]. Необхідно відзначити, що принцип адитивності може бути застосований для разрахунку зміни стійкості комплексу праймер одноланцюгова ДНК у біля температури плавлення, але він вимагає корекції ентальпії й ентропії. Необхідність корекції ентальпії й ентропії пов'язана з тим, що ентальпія й ентропія залежать від температури рівняння (1.6) і (1.7).
![]() (1.6)
(1.6)
![]() (1.7)
(1.7)
де ![]() - зміна теплоємності в ході реакції [60].
- зміна теплоємності в ході реакції [60].
Вплив ![]() на температуру плавлення описується рівнянням (1.8) [62].
на температуру плавлення описується рівнянням (1.8) [62].
![]() (1.8)
(1.8)
де: R - універсальна газова постійна.
На малюнку № 1.11. представлена залежність фракції двухланцюгової ДНК від температури з різною часткою АТ основ [54]. Отримані теоретичні дані близькі до практичних (рис. № 1.10.). Однак принцип адитивності не враховує взаємодій між сусідніми нуклеотидами. Це призводить до відсутності залежності між довжиною двухланцюгового олігонуклеотиду і температурою його плавлення, що суперечить практично отриманим даним. Продемонструємо це на простому прикладі. Припустимо в нас є ряд двухланцюгових олігонуклеотидів, що складаються з однакових нуклеотидів. Тоді ![]() ,
, ![]() і
і ![]() . Так як температура плавлення - це та температура, при якій
. Так як температура плавлення - це та температура, при якій ![]() , то
, то ![]() . Отриманий вираз не відповідає даним експерименту, який свідчить що чим більша довжина олігонуклеотида, тим вище температура його плавлення.
. Отриманий вираз не відповідає даним експерименту, який свідчить що чим більша довжина олігонуклеотида, тим вище температура його плавлення.
Висновок: існують додаткові взаємодії, які не враховані принципом адитивності.
Для урахування впливу послідовності нуклеотидів на стійкість ДНК розроблена модель (Nearest Neighbor Model), яка припускає, що на кожний нуклеотид впливає два його сусіди. На підставі емпіричних досліджень розраховані ентальпії й ентропії для всіх пар основ, що приведені в таблиці № 1.10.
Таблиця 1.10. Термодинамічні параметри для nearest-neighbor melting point formula.
|
Взаємодія |
ДНК |
РНК |
||
|
ККал/моль |
ККал/К.моль |
ККал/моль |
ККал/К.моль |
|
|
AA/TT |
-9.1 |
-24.0 |
-6.6 |
-18.4 |
|
AT/TA |
-8.6 |
-23.9 |
-5.7 |
-15.5 |
|
TA/AT |
-6.0 |
-16.9 |
-8.1 |
-22.6 |
|
CA/GT |
-5.8 |
-12.9 |
-10.5 |
-27.8 |
|
GT/CA |
-6.5 |
-17.3 |
-10.2 |
-26.2 |
|
CT/GA |
-7.8 |
-20.8 |
-7.6 |
-19.2 |
|
GA/CT |
-5.6 |
-13.5 |
-13.3 |
-35.5 |
|
CG/GC |
-11.9 |
-27.8 |
-8.0 |
-19.4 |
|
GC/CG |
-11.1 |
-26.7 |
-14.2 |
-34.9 |
|
GG/CC |
-11.0 |
-26.6 |
-12.2 |
-29.7 |
|
Initiation |
0.0 |
-10.8 |
0.0 |
-10.8 |
Температура плавлення олігонуклеотиду згідно NN моделі може бути розрахована згідно рівняння (1.6).
![]() (1.6)
(1.6)
де: ![]() - сумарна зміна ентальпії, A – малий, але важливий коригувальний коефіцієнт утворення подвійного ланцюга,
- сумарна зміна ентальпії, A – малий, але важливий коригувальний коефіцієнт утворення подвійного ланцюга, ![]() - сума зміни NN ентропії, R – універсальна газова постійна (1,99 кал/К.моль), C – концентрація олігонуклеотидів,
- сума зміни NN ентропії, R – універсальна газова постійна (1,99 кал/К.моль), C – концентрація олігонуклеотидів, ![]() - концентрація катіона
- концентрація катіона ![]() .
.
Після відпалювання праймерів Taq-полімераза починає добудовування другого ланцюга ДНК із 3'-кінця праймера.
3. Елонгація ланцюга. На третьому етапі температуру в реакційній суміші доводять до оптимуму роботи Taq-полімерази, і синтез другого ланцюга продовжується з максимальною ефективністю. Розповсюджене використання температури елонгації ланцюга 72ºС тому, що на цій температурі полімеразна активність максимальна. Залежність активності полімерази від температури приведена в таблиці № 1.5. Активність визначається по включенню мітки в денатуровану і відпалену salmon sperm ДНК, взяту у великому надлишку.
Однак, температури елонгації ланцюга 72ºС прийнятна не для всіх випадків:
1. В описі експерименту для визначення активності полімерази є важливий момент: матриця взята у великому надлишку, тобто, полімераза має можливість "зневажати" незручні ділянки синтезу ДНК. В реальному процесі полімеразі необхідно пройти всі ділянки ланцюга. Виявляється, що при температурі ~65ºС синтез менше залежить від характеру матриці, що особливо актуально при синтезі GC-багатого фрагмента, (але навіть ця температура занадто велика для буфера з бетаіном).
2. Матриці зі змістом ~90% А-Т основ вдається ампліфікувати тільки знизивши температури елонгації ланцюга до температури ~60ºС (при цьому швидкість синтезу знижується до 1kb/хв.) [44].
Таблиця 1.11.
|
Температура ºС |
Швидкість синтезу |
|
|
нукл/сек |
kb/хв. |
|
|
75-80 |
~150 |
~9 |
|
70 |
>60 |
>3.6 |
|
55 |
24 |
1.4 |
|
37 |
1.5 |
190 |
|
22 |
0.25 |
15 |
У випадку близького значення температури відпалювання праймерів і температури оптимуму роботи ферменту, стає можливим використовувати двухетапний ПЛР, сполучивши відпалювання та елонгацію.
Температурний цикл ампліфікації багаторазово повторюється (30 і більше разів). На кожному циклі кількість синтезованих копій фрагмента ДНК подвоюється.
Результатом циклічного процесу є експонентне збільшення кількості специфічного фрагмента ДНК, яке можна описати формулою (1.7).
![]() ~
~ ![]() , (1.7)
, (1.7)
де: А - кількість специфічних продуктів реакції ампліфікації;
М – початкова кількість ДНК-мішеней;
n - число циклів ампліфікації.
Реальне значення ефективності окремих циклів ампліфікації складає 78-97%. У випадку присутності в пробі інгібіторів реакції це значення може бути набагато менше, тому фактична кількість специфічних продуктів ампліфікації краще описує рівняння (1.8).
![]() , (1.8)
, (1.8)
де: Е -ефективність реакції.
Варто помітити, що в процесі ампліфікації на вихідному ланцюзі синтезуються і довгі фрагменти, однак їх нагромадження відбувається лише в арифметичній прогресії по формулі (1.9).
![]() , (1.9)
, (1.9)
де K – кількість довгих продуктів ампліфікації.
Таким чином, специфічні фрагменти, обмежені на кінцях праймерами, вперше з'являються наприкінці другого циклу, накопичуються в геометричній прогресії і незабаром починають домінувати серед продуктів ампліфікації.
4. Цикли після ПЛР. Для перетворення деякої залишкової кількості одноланцюгових матриць у двухланцюгові продукти рекомендуеться проводити додаткові цикли без денатурації: тільки відпалювання та подовження.
1.7.3. Кількість циклів ампліфікації.
Чим більше кількість ПЛР-циклів, тим вища чутливість методу і нижча специфічність. Однак максимальної чутливості не можна досягти тільки збільшенням числа циклів, необхідно оптимізувати інші параметри, для того щоб підвищити ефективність кожного циклу. Кількість циклів ампліфікації залежить також від змісту в зразку потрібної послідовності. Для виявлення унікальних генів, які присутні у кожній клітці (наприклад, при диагностиці спадкоємних захворювань), то досить 20 - 30 циклів. Якщо ж мішень знаходиться тільки в частині кліток досліджуваного препарату (наприклад, у випадку вірусної інфекції), то кількість циклів може складати 30 - 50.
Необхідно відзначити, що при збільшенні числа циклів вихід продукту проходить три фази:
1. Вихід продукту збільшується в геометричній прогресії;
2. Вихід продукту стабілізується в наслідку «Ефекту плато», що розглянутий у п. 1.7.2.4.
3. Зниження концентрації ПЛР-продукту [57].
На малюнку № 1.11. приведений знімок електрофорезу ПЛР продуктів, що були отримані при ампліфікації двох зразків ДНК із варіюванням числа циклів ампліфікації від 21 до 45, із кроком 3 цикли [57].
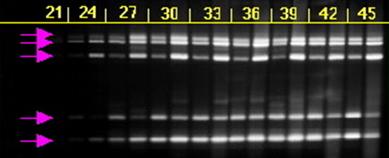
Рис. 1.11.
1.7.4. Ефект плато при збільшенні кількості ПЛР циклів.
Варто помітити, що процес нагромадження специфічних продуктів ампліфікації по геометричній прогресії йде лише обмежений час, а потім його ефективність критично падає. Це зв'язано з так званим «ефектом плато».
Термін «ефект плато» використовують для опису процесу накопичення продуктів ПЛР на останніх циклах ампліфікації, коли кількість ампліконів досягає 0,3 - 1 пмолей.
В залежності від умов і кількості циклів реакції ампліфікації, на момент досягнення «ефекту плато» впливають:
- утилізація субстратів (дНТФ і праймерів);
- стабільність реагентів (дНТФ і ферменту);
- кількість інгібіторів, включаючи пірофосфати і ДНКи-дуплекси;
- неспецифічні продукти або праймер-димери, що конкурують за праймери, дНТФ і полімеразу;
- концентрація специфічного продукту;
- неповна денатурація при високій концентрації продуктів ампліфікації.
Чим меньше початкова концентрація ДНК-мішені, тим вище ризик виходу реакції на плато. Цей момент може наступити до того, як кількість специфічних продуктів ампліфікації буде достатньою, щоб їх можна було проаналізувати. Уникнути цього дозволяють лише добре оптимізовані тест-системи.
1.7.5. Модифікації постановки ПЛР
Полімеразна ланцюгова реакція з використанням «гарячого старту».
Щоб зменшити ризик утворення неспецифічних продуктів реакції ампліфікації, використовують підхід, що одержав назву «гарячий старт» (від англ. «hot-start»). Суть його полягає в запобіганні можливості початку реакції до моменту досягнення в пробірці умов, що забезпечують специфічний віджиг праймерів.
Справа в тому, що в залежності від ГЦ-складу і розміру, праймери мають певну температуру плавлення (Tm), при якій утворення водневих зв'язків нестабільно. Якщо температура системи перевищує Тм, праймер не в змозі утримуватися на ланцюзі ДНК і денатурує. При дотриманні оптимальних умов, тобто температури відпалювання близької до температури плавлення, праймер утворить двухланцюгову молекулу тільки за умови його повної комплементарності і, таким чином, забезпечує специфічність реакції.
Існують різні варіанти реалізації «гарячого старту»:
1. внесення в реакційну суміш Taq-полімерази під час першого циклу після прогріву пробірки до температури денатурації;
2. розподіл інгредієнтів реакційної суміші прошарком, наприклад, парафіну або воску на частини (у нижній - праймери, у верхній - Taq-полімераза і ДНК-мішені), що змішуються при досягненні температури плавлення матеріалу прошарку (~45-85ºС);
3. використання моноклональних антитіл до Taq-полімерази. Фермент, зв'язаний моноклональними антитілами, стає активним лише після стадії першої денатурації, коли моноклональні антитіла незворотньо денатурують, і звільняють активні центри Taq-полімерази.
В усіх вказаних випадках, навіть якщо неспецифічний віджиг відбувся до початку температурного циклу, елонгації не відбувається, а при нагріванні комплекси праймер-ДНК денатурують, тому неспецифічні продукти не утворяться. Надалі температура в пробірці не опускається нижче температури плавлення, що забезпечує утворення специфічного продукту ампліфікації.
«Nested» ПЛР.
Одним з засобів підвищити чутливість реакції є застосування методу «nested» ампліфікації. Суть його полягає в послідовному використанні двох пар праймерів – зовнішньої і внутрішній. Після використання першої пари праймерів продукт ампліфікації переносять в іншу пробірку з внутрішньою парою праймерів. Іноді замість «nested» ампліфікації використовують процес реампліфікації. У цьому випадку проводять додатковий раунд ампліфікації, у якому використовують колишню пару праймерів, а як ДНК-мішень – продукт першої реакції ампліфікації.
Така схема постановки ампліфікації більш трудомістка, оскільки, як правило, робить необхідним постановку двох реакцій ампліфікації замість однієї і вимагає особливо ретельної облаштованості лабораторних приміщень, яка дозволяє гарантированно уникати контамінації продуктами реакції після використання зовнішньої пари праймерів. До «гнездной» ампліфікації або реампліфікації прибігають лише в особливих випадках, тому що сучасні ПЛР-набори дозволяють добиватися тих же результатів іншими засобами.
Напівкількісний аналіз
Для більш точної оцінки кількості ДНК-мішені в реакційній суміші використовуються спеціальні підходи, відомі під загальною назвою напівкількісного ПЛР-аналізу. Префікс «напів» має принципове значення через умовну точність результатів цього аналізу.
До таких підходів варто віднести використання спеціального приладового забезпечення в сполученні з препаратами специфічної ДНК із відомою концентрацією.
До приладового забезпечення, що дозволяє одержувати напівкількісні результати ПЛР аналізу, варто віднести моделі «iCycler IQtm» (Био-Рад, США) і «COBAS Amplicor» (Roche, США), що дозволяють стежити за кінетикою нагромадження продуктів ампліфікації. Це досягається рішенням на стику фізики і біології. У реакційну суміш уводять гібридизаційні зонди, до складу яких входять нуклеотиди, помічені особливими реактивами – флуорофором і тушителем. Флуорофори здатні випромінювати енергію лише у вільному від тушителя стані. На стадії відпалювання відбувається гібридизація зондів із внутрішніми ділянками ампліконів. У процесі елонгації Taq полімераза руйнує зонди за рахунок своєї екзонуклеазної активності. Це приводить до влучення мічених нуклеотидів у розчин, де вони починають флуоресцировати (рис. 1.12.). Інтенсивність флуоресценсії фіксується спеціальним детектором і є пропорційною кількості продуктів ампліфікації. Якщо гібридизація не проходить, то зонд залишається цілим, а флуорофори, що входять у його склад, не дають випромінювання.
При дослідженні зразків кожна серія експериментів супроводжується постановкою ампліфікації з контрольними зразками, у яких свідомо відоме кількість копій ДНК. Порівняння кінетики нагромадження продуктів ампліфікації в реакційних сумішах в експериментальному і контрольному зразках дозволяє напівкількісно оцінити концентрацію ДНК у діапазоні розведень контрольних препаратів ДНК.
Більш простим, але менш надійним варіантом оцінки кількості специфічної ДНК є метод розведень. Детекцію проводять після гібридизації з ДНК-зондами, чи методом електрофорезу, визначаючи кількість працюючих розведень.
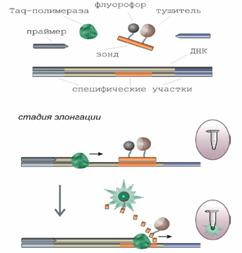
Рис. 1.12. Використання флуоресцентних зондів у процесі ампліфікації.
При всій привабливості напівкількісного аналізу слід зазначити, що для його виконання допускається використання тільки препаратів ДНК із високим ступенем очищення. Важливо відзначити, що існують домішки, що можуть по-різному впливати на ефективність ампліфікації досліджуваної і контрольної ДНК. При використанні спрощених методів виділення ДНК у більшості випадків не представляється можливим заздалегідь пророчити кількість і склад домішок у клінічних зразках.
Відомо, що в деяких випадках можливі втрати ДНК на стадії виділення, що може істотно спотворити значення реальної кількості ДНК у зразку.
Крім того, точне знання кількості збудників у конкретному клінічному зразку далеко не завжди може дати повну уяву про інфекційний процес, наприклад, через різну локалізацію і вірулентність мікроорганізмів, а також стану імунної системи організму хазяїна.
Таким чином, існуючі варіанти напівкількісного аналізу поки ще не мають великої практичної цінності для рутинного клінічного тестування. У той же час їхня користь незаперечна при оцінці ефективності терапії при деяких інфекційних захворюваннях.
1.7.6. Контроль за проходженням реакції ампліфікації
Результат ПЛР можна кваліфікувати як позитивний або негативний у залежності від того, виявлена в зразку цікавляча нас послідовність-мішень чи ні. Однак порушення нормального ходу ампліфікації, недостатня чутливість методу і непередбачений поліморфізм послідовності-мішені в області зв'язування праймерів або гібридизаційного зонда може дати хибнонегативний результат. У випадку забруднення зразків і випадкової гомології між зондом, праймерами і послідовністю, подібної з мішенню, виходять хибнопозитивні результати. Проблеми, що виникають у зв'язку з перехресними реакціями за участю зонда або праймерів і можливим поліморфізмом.
Дуже важливим для правильної інтерпретації результатів є вибір контролів. Позитивні і негативні контролі повинні бути добре охарактеризовані. Часто використовують ДНК із клітинних ліній, що свідомо містять чи не містять послідовність-мішень. У кожному аналізі потрібні як мінімум три контролі:
• позитивний контроль;
• негативний контроль;
• бланк-контроль.
Бланк-контроль - це реакційна суміш, у якій присутні усі компоненти, за винятком ДНК; він є індикатором забруднень. Один тип позитивного контролю повинний містити максимальну кількість послідовностей-мішеней, іншої - невелику їх кількість. Це дозволяє визначити чутливість і ефективність ПЛР.
У деяких випадках ампліфікація взагалі не йде, і якщо є підстави думати, що отриманий результат хибнонегативний, дуже важко визначити, по якій саме причині. Щоб вирішити цю проблему, кожен зразок необхідно тестувати, проводячи ампліфікацію якої-небудь геномної послідовності, наприклад гена рецептора ліпопротеінів низкої щільності або ß-глобінового гена. При цьому розмір геномної послідовності-мішені повинн бути трохи більше, ніж досліджуваної; це дозволить переконатися, що ДНК зразка не занадто сильно деградована. При ПЛР будь-якого зразка тканин людини геномна ДНК повинна ампліфікуватися. Відсутність ампліфікації може означати інгибування ферменту, сильну деградацію ДНК або те, що її занадто мало. У цьому випадку треба повторити ампліфікацію зразка, збільшивши і зменшивши кількість ДНК-матриці або збільшивши концентрацію ДНК-полімерази Tag. Ампліфікація може бути відсутньою, незважаючи на всі зусилля. Це може бути пов'язане із сильним некрозом досліджуваної тканини або фіксацією препарату не в 10% формаліні, а в інших фіксаторах. У цьому випадку єдиним виходом є повторна біопсія.
Геномні праймери можна використовувати в одному раунді ПЛР разом із праймерами, специфічними у відношенні послідовності-мішені: ампліфікація двох послідовностей йде при цьому одночасно. Такий множинний ПЛР-тест може бути менш чуттєвим, чим той, у якому використовується один набір праймерів, але він виключає появу хибнонегативного результату [44].
1.8. Виділення ДНК матриці
При підготовці проби до постановки ПЛР використовують різні методики в залежності від поставлених задач. Їхня суть полягає в екстракції (витягу) ДНК із біопрепарату і видаленні або нейтралізації сторонніх домішок для одержання препарату ДНК із чистотою, придатною для постановки реакції ампліфікації.
Іноді буває досить прокип'ятити зразок протягом 5-10 хвилин, однак у більшості випадків вимагаються більш трудомісткі методи.
Стандартною і ставшою уже класичною вважається методика одержання чистого препарату ДНК, запропонована Marmur. Вона містить у собі ферментативний протеоліз кліток з наступної депротеінізацією і переосадженням ДНК спиртом. Однак цей метод є досить трудомістким і припускає роботу з такими агресивними речовинами, що мають різкий запах, як фенол і хлороформ.
Одним з популярних у даний час є метод виділення ДНК, запропонований Boom із співавторами. Цей метод заснований на використанні для лізису кліток сильного хаотропного агента - гуанідіна тіоционата (GuSCN) і наступної сорбції ДНК на носії (скляне намисто, діатомова земля, скляне «молоко» і т.п.). Після відмивань у пробі залишається ДНК, сорбованая на носії, з якого вона легко знімається за допомогою елюіруючого буфера. Метод зручний, технологичний і придатний для підготовки зразка до ампліфікації. Однак можливі втрати ДНК унаслідок необоротної сорбції на носії, а також у процесі численних відмивань. Особливо велике значення це має при роботі з невеликими кількостями ДНК у зразку. Крім того, навіть слідові кількості GuSCN можуть інгібіровати ПЛР. Тому при використання цього методу дуже важливим є правильний вибір сорбенту і ретельне дотримання технологічних нюансів. Слід зазначити, що через велику кількість стадій додавання і видалення розчинів при роботі зі зразком потрібна акуратність, тому що можлива перехресна контамінація між пробами що утвориться аерозолью ДНК.
Інша група методів пробопідготування заснована на використанні іонообмінників типу Chilex, що, на відміну від скла, сорбують не ДНК, а, навпаки, домішки, що заважає реакції. Як правило, ця технологія включає дві стадій: кип'ятіння зразка, у результаті чого клітинні стінки руйнуються, а нуклеінові кислоти виходять у розчин; і сорбція домішок на іонообміннику. Метод надзвичайно привабливий простотою виконання. У більшості випадків він придатний для роботи з клінічним матеріалом. На жаль, іноді зустрічаються зразки з такими домішками, що неможливо видалити за допомогою іонообмінників. Крім того, клітинні стінки деяких мікроорганізмів не піддаються руйнуванню простим кип'ятінням. У цих випадках є необхідним вводити додаткові стадії обробки зразка.
При масовому скринінгу, коли важливо одержати статистичні дані, допускається використання простих методів із застосуванням детергентів або обробки біологічного матеріалу лугами з наступною їх нейтралізацією. У той же час, використання подібних методів пробопідготування для клінічної діагностики може приводити до хибнонегативних результатів унаслідок використання в реакційній суміші неякісного препарату ДНК.
Таким чином, до вибору методу пробопідготування варто ставитися з розумінням цілей проведення передбачуваних аналізів.
1.9. Детекція продукту полімеразної ланцюгової реакції
Для правильної оцінки результатів ПЛР важливо розуміти, що даний метод не є кількісним. Теоретично продукти ампліфікації одиничних молекул ДНК-мішені можуть бути виявлені за допомогою електрофорезу вже після 30-35 циклів. Однак на практиці це виконується лише у випадках, коли реакція проходить в умовах, близьких до ідеальних, що в житті зустрічається не часто. Особливо великий вплив на ефективність ампліфікації надає ступінь чистоти препарату ДНК, тобто наявність у реакційній суміші тих чи інших інгібіторів, від яких позбутися в деяких випадках буває вкрай складно. Іноді через їх присутність не вдається ампліфікувати навіть десятки тисяч молекул ДНК-мішені. Таким чином, прямий зв'язок між вихідною кількістю ДНК-мішені і кінцевою кількістю продуктів ампліфікації часто відсутній [44].
Метод горизонтального електрофорезу. Для візуалізації результатів ампліфікації використовують різні методи. Найбільш розповсюдженим на сьогоднішній день є метод електрофорезу, заснований на розділі молекул ДНК за розміром. Для цього готують пластину агарозного гелю, що представляє собою застиглу після розплавлювання в електрофорезному буфері агарозу в концентрації 1,5-2,5% з додаванням спеціального барвника ДНК, - наприклад, бромістого етидія. Застигла агароза утворить просторові ґрати. При заливанні за допомогою гребінок у гелі формують спеціальні лунки, у які надалі вносять продукти ампліфікації. Пластину гелю містять в апарат для горизонтального гель-електрофорезу і підключають до джерела постійної напруги. Негативно заряджена ДНК починає рухатися в гелі від мінуса до плюса. При цьому більш короткі молекули ДНК рухаються швидше, ніж довгі. На швидкість руху ДНК у гелі впливає концентрація агарози, напруженість електричного поля, температура, склад електрофорезного буфера і, у меншому ступені, Гц-склад ДНК. Усі молекули одного розміру рухаються з однаковою швидкістю. Барвник убудовується (інтеркалірує) площинними групами в молекули ДНК. Після закінчення електрофорезу, що продовжується від 10 хв до 1 години, гель поміщають на фільтр трансілюмінатора, що випромінює світло в ультрафіолетовому діапазоні (254, 310 нм). Енергія ультрафіолету, що поглинається ДНК в області 260 нм, передається на барвник, змушуючи його флуоресцировати в оранжево-червоній області видимого спектру (590 нм).
Яскравість смуг продуктів ампліфікації може бути різною. Тому часто в ПЛРх-лабораторіях прийнято оцінювати результат по трьох-, чотирьох- чи п'ятибальній системі. Однак, як уже відзначалося раніше, це не можна зв'язувати з початковою кількістю ДНК-мішені в зразку. Часте зменшення яскравості світіння смуг пов'язано зі зниженням ефективності ампліфікації під впливом інгібіторів або інших факторів.
Метод вертикального електрофорезу. Метод вертикального електрофорезу принципово схожий з горизонтальним електрофорезом. Їх відмінність полягає в тому, що в даному випадку замість агарози використовують поліакріламід. Його проводять у спеціальній камері для вертикального електрофорезу. Електрофорез в поліакриламидному гелі має велику розділяючу здатність у порівнянні з агарозним електрофорезом і дозволяє розрізнювати молекули ДНК різних розмірів з точністю до одного нуклеотиду. Готування поліакриламідного гелю трохи складніше агарозного. Крім того, акриламід є токсичною речовиною. Оскільки необхідність визначити розмір продукту ампліфікації з точністю до 1 нуклеотиду виникає рідко, то в рутинній роботі цей метод не використовують.
1.10. Проблема контамінації
Потенційно висока чутливість полімеразної ланцюгової реакції робить зовсім необхідним особливо ретельний пристрій ПЛР-лабораторії. Це пов'язано з найбільш гострою проблемою методу – контамінацією.
Контамінація – влучення з зовнішнього середовища в реакційну суміш специфічних і неспецифічних молекул ДНК, здатних служити мішенями в реакції ампліфікації і давати хибнопозитивні чи хибнонегативні результати.
Такими мішенями можуть бути продукти реакції, що попадають у зовнішнє середовище на етапі детекції з пробірок, у яких успішно пройшла ампліфікація, або специфічна ДНК зі зразків на етапі пробопідготування.
Існує кілька способів боротьби з цим неприємним явищем. Одним з них є використання ферменту N-урацил-гликозілази (УГ). В основі цього методу лежить здатність УГ розщеплювати молекули ДНК з убудованим урацилом. Реакцію ампліфікації проводять з використанням суміші дНТФ, у якій дТТФ замінений на дУТФ (урацил), і після термоциклірування всі амплікони, що утворяться в пробірці, будуть містити урацил. Якщо до ампліфікації в реакційну суміш додати УГ, то амплікони, що потрапили в реакційну суміш, будуть зруйновані, тоді як нативна ДНК залишиться цілою і буде надалі служити мішенню для ампліфікації.
Іншим способом інактивації ампліконів служить фотохімічний вплив на молекули ДНК. Для цього використовують псорален чи ізопсорален, що активується короткочасним опроміненням ультрафіолетовим світлом. Модифіковані цими з'єднаннями молекули ДНК не можуть брати участь у реакції ампліфікації.
Однак, як відомо, жодна біологічна чи хімічна реакція не йде з 100% ефективністю і, відповідно, після інактивації продуктів ампліфікації з мільярдів копій ампліфіцированого фрагменту хоча б трохи залишиться цілим. Це істотно знижує цінність такого підходу. Крім того, завжди залишається ризик кроса-контамінації від зразка до зразка в процесі пробопідготування.
Таким чином, обоє ці методи лише до деякої міри дозволяють усунути джерело контамінації і не гарантують від хибнопозитивних і хибнонегативних результатів.
Для впевненості у відсутності контамінації необхідно кожну серію експериментів супроводжувати негативними контролями. Як негативні контролі рекомендується використовувати воду з кімнати пробопідготування.
Усі реактиви рекомендується зберігати розлитими на окремі порції (аліквоти).
